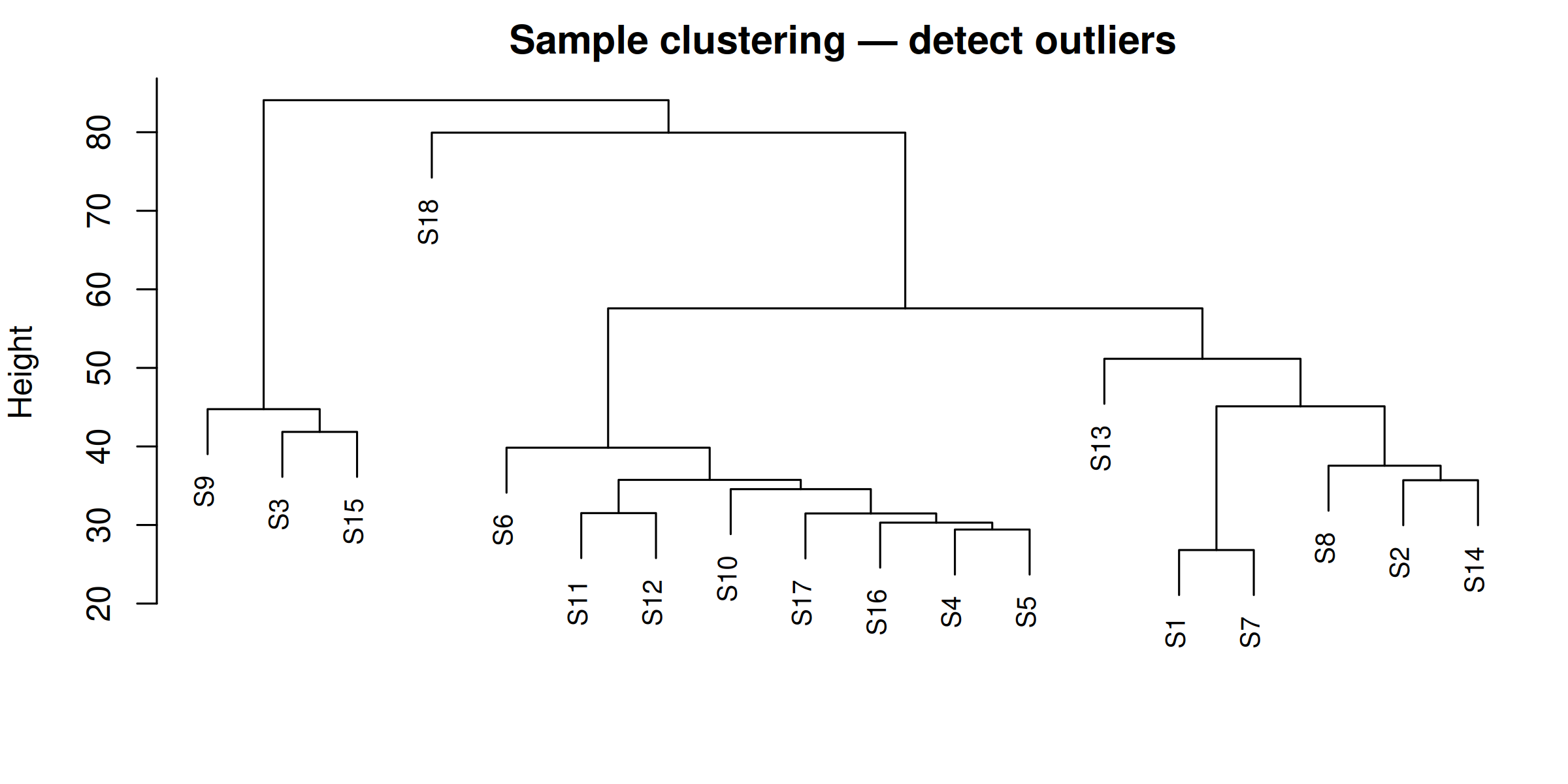
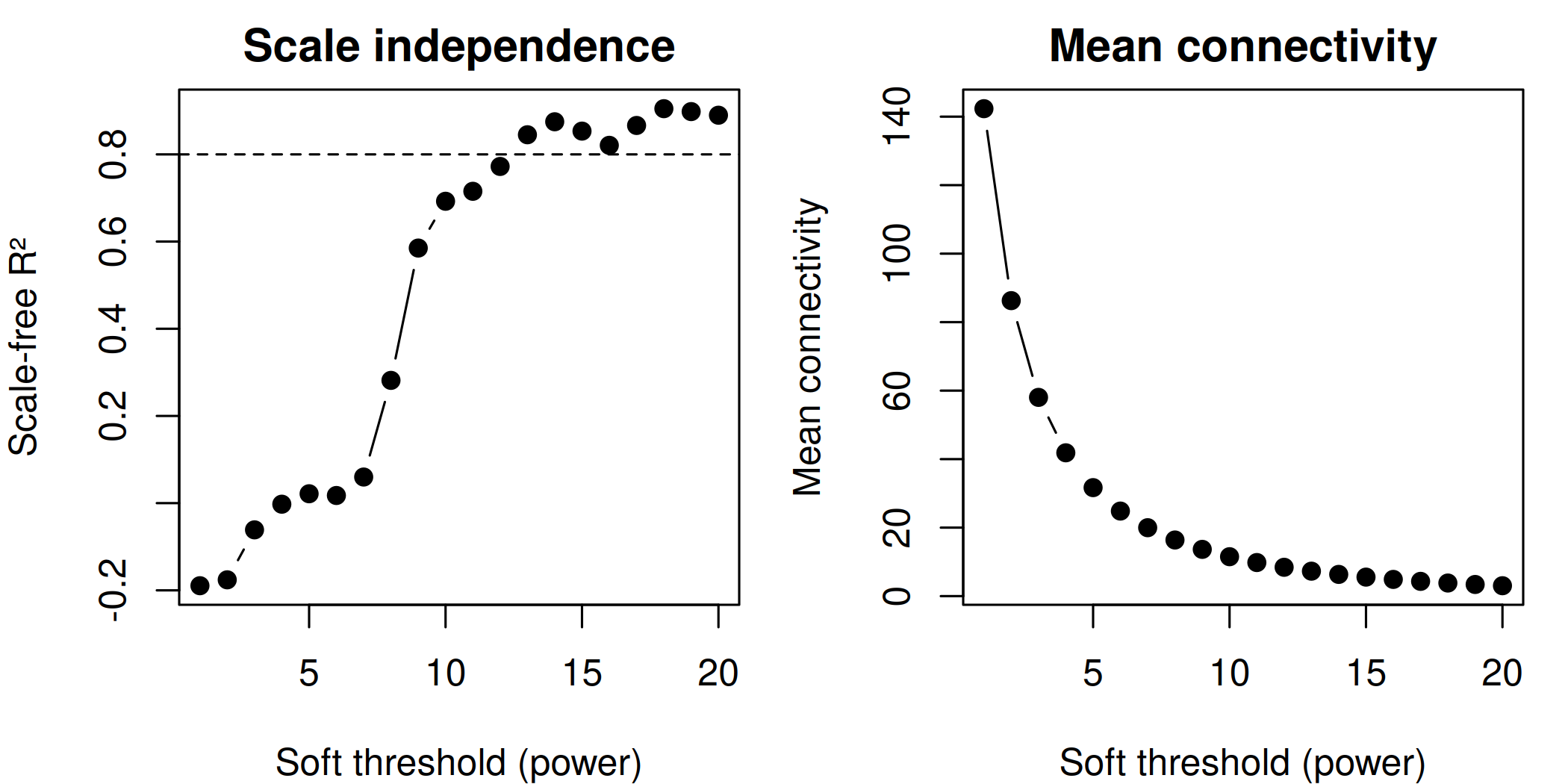
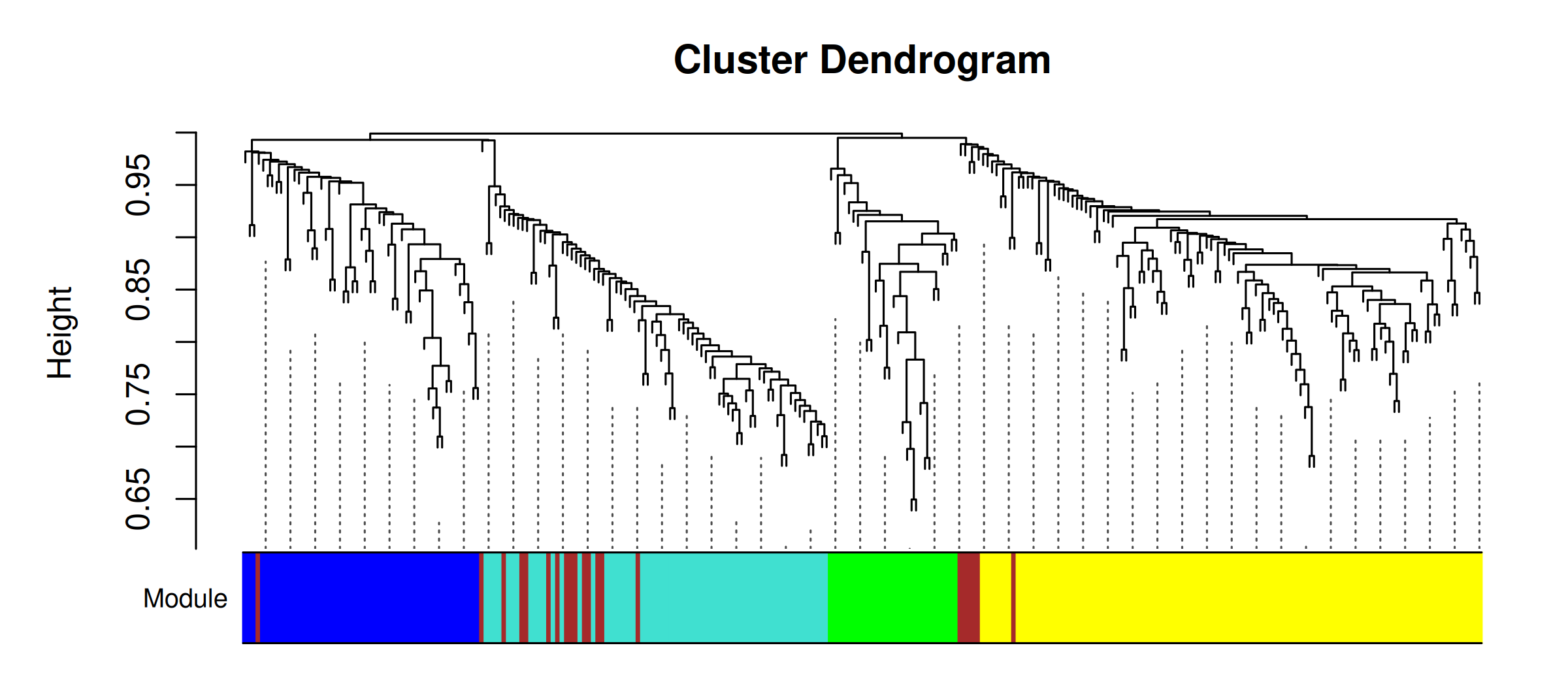
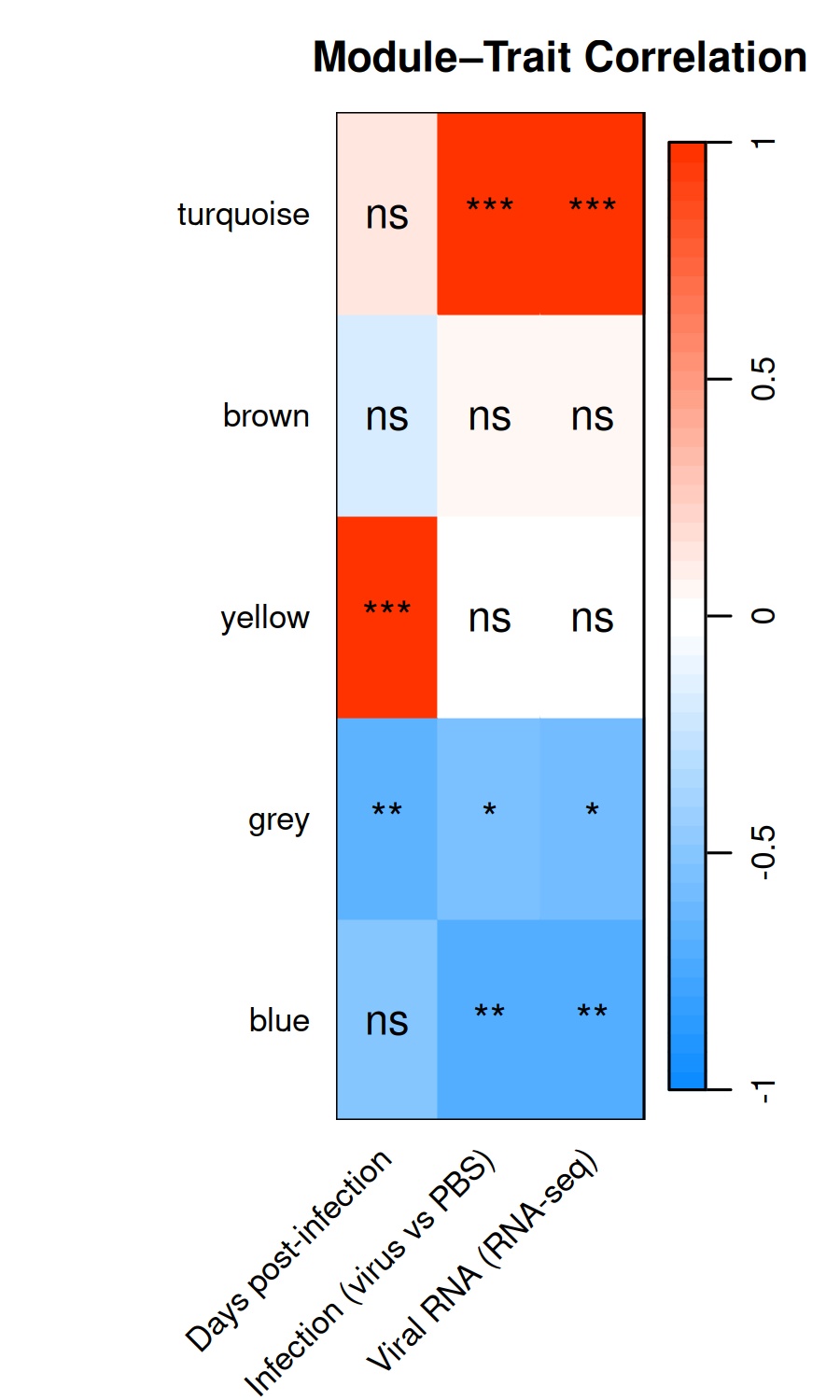
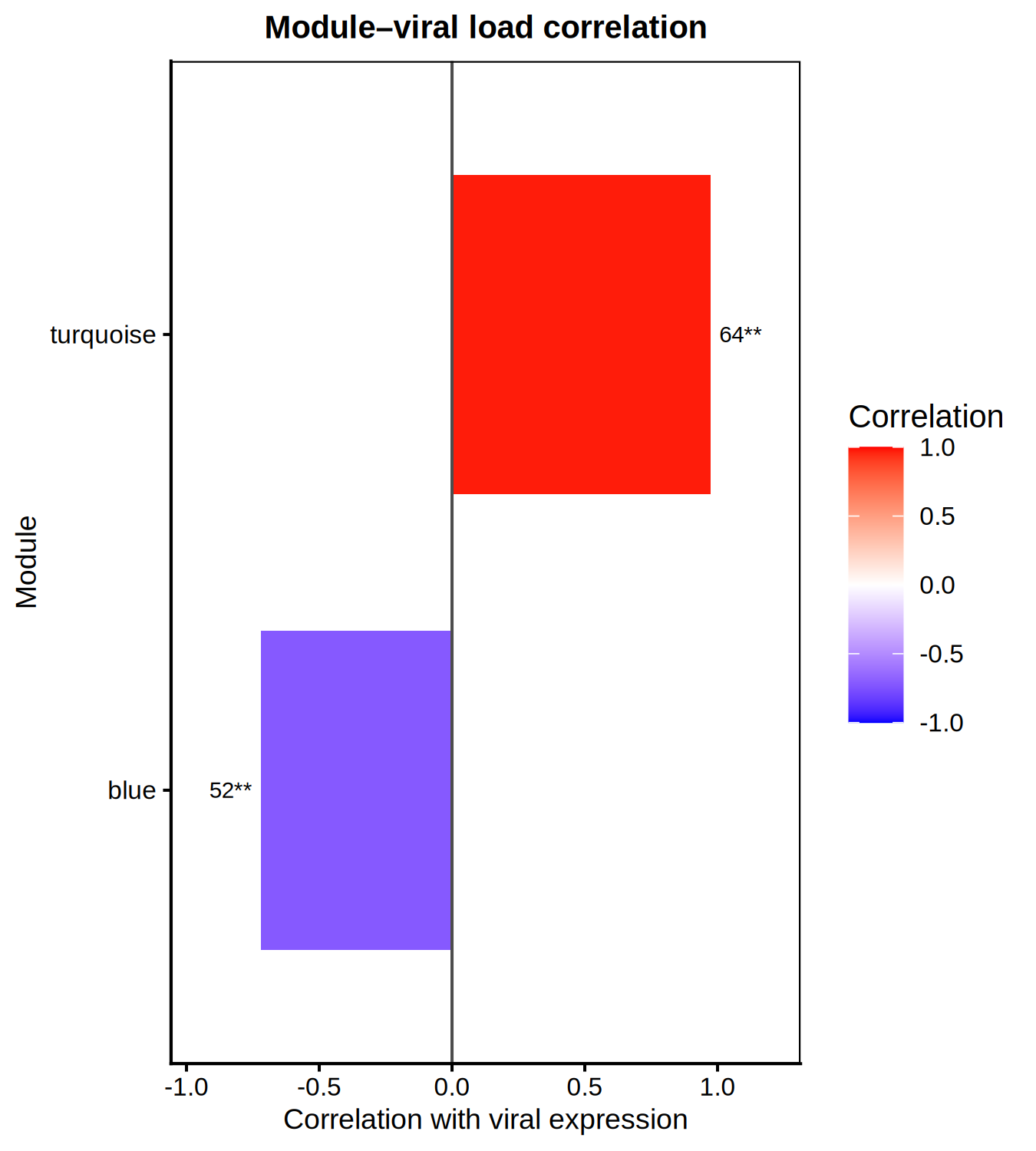
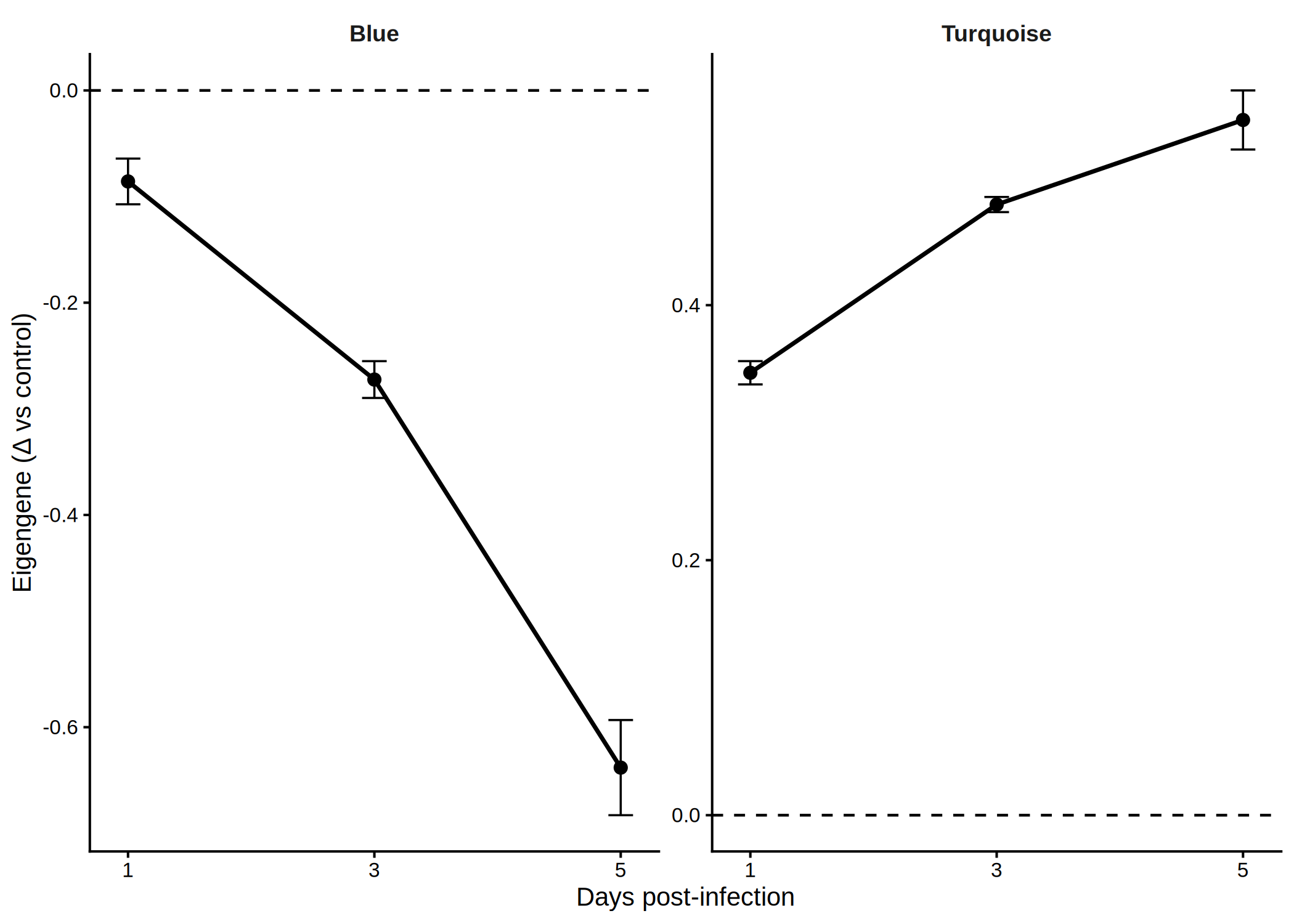
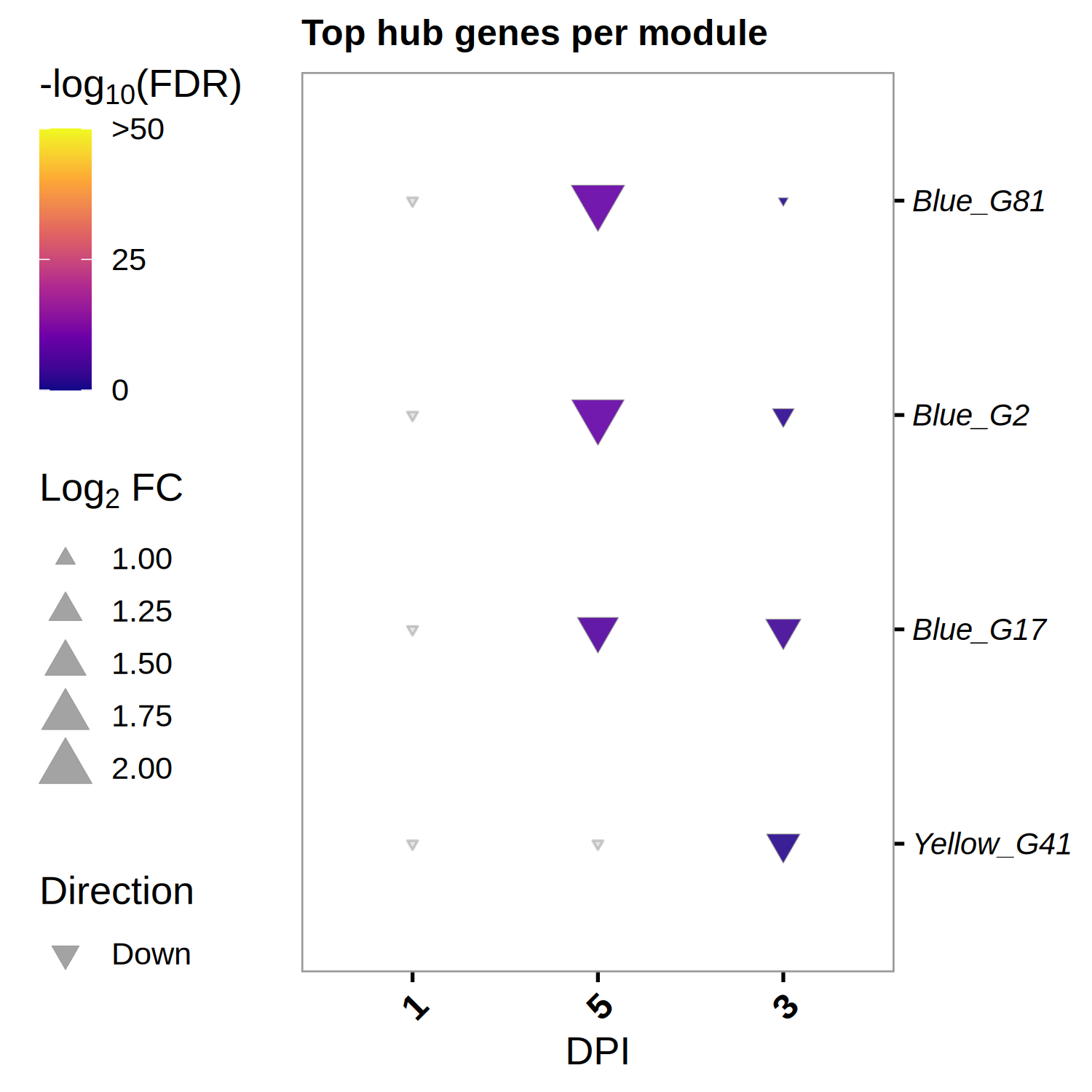
# Top 3 hub genes per module (from DEG set) <- hub_tbl %>% :: filter (Gene %in% DEG_genes) %>% :: arrange (Module, dplyr:: desc (hubness)) %>% :: group_by (Module) %>% :: slice_head (n = 3 ) %>% :: ungroup () %>% :: select (Gene, Module)if (nrow (top3) == 0 ) {message ("No hub genes overlap with DEGs — all genes will be shown." )<- hub_tbl %>% :: arrange (Module, dplyr:: desc (hubness)) %>% :: group_by (Module) %>% :: slice_head (n = 3 ) %>% :: ungroup () %>% :: select (Gene, Module)<- top3 %>% dplyr:: arrange (Module, Gene) %>% dplyr:: pull (Gene) %>% unique ()<- de_long %>% :: inner_join (top3, by = "Gene" ) %>% :: mutate (Comparison = factor (as.character (dpi), levels = as.character (sort (unique (dpi)))),Gene = factor (Gene, levels = ordered_genes),color_group = ifelse (! is.na (padj) & padj < 0.05 , "significant" , "nonsignificant" ),log_padj = ifelse (! is.na (padj) & padj > 0 , - log10 (padj), 0 ),direction = ifelse (log2FoldChange > 0 , "Up" , "Down" )<- 50 <- ggplot () + geom_point (data = dplyr:: filter (plot_bubble, color_group == "nonsignificant" ),aes (x = Comparison, y = Gene),shape = 25 , size = 0.8 , color = "grey60" , fill = "grey80" , alpha = 0.5 + geom_point (data = dplyr:: filter (plot_bubble, color_group == "significant" ),aes (x = Comparison, y = Gene,size = abs (log2FoldChange),fill = pmin (log_padj, cap_val),shape = direction),color = "grey60" , stroke = 0.2 , alpha = 0.9 + scale_shape_manual (values = c (Up = 24 , Down = 25 ), name = "Direction" ,guide = guide_legend (override.aes = list (fill = "grey60" , size = 3 ))) + scale_size_continuous (name = expression ("Log" [2 ] * " FC" ),guide = guide_legend (override.aes = list (shape = 24 , fill = "grey60" ))) + scale_fill_viridis_c (option = "plasma" , name = expression ("-log" [10 ] * "(FDR)" ),limits = c (0 , cap_val), breaks = c (0 , 25 , cap_val),labels = c ("0" , "25" , paste0 (">" , cap_val))+ scale_y_discrete (position = "right" , drop = FALSE ) + labs (title = "Top hub genes per module" , x = "DPI" , y = NULL ) + theme_classic (base_size = 13 , base_family = "Arial" ) + theme (axis.text.x = element_text (size = 12 , angle = 45 , hjust = 1 , face = "bold" ),axis.text.y = element_text (size = 10 , face = "italic" ),legend.position = "left" ,panel.border = element_rect (color = "grey60" , fill = NA , linewidth = 0.6 ),axis.line = element_blank (),plot.title = element_text (size = 12 , face = "bold" )ggsave ("Plots/WGCNA_HubGene_Bubble.tiff" ,width = 6 , height = 5 , dpi = 300 , compression = "lzw" )ggsave ("Plots/WGCNA_HubGene_Bubble.pdf" ,width = 6 , height = 5 , device = cairo_pdf)print (p_bubble)