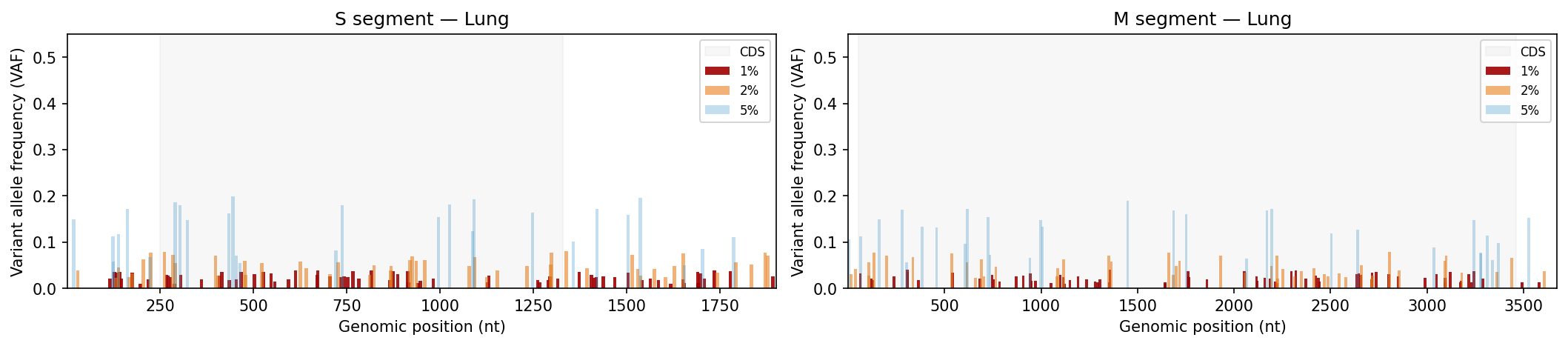
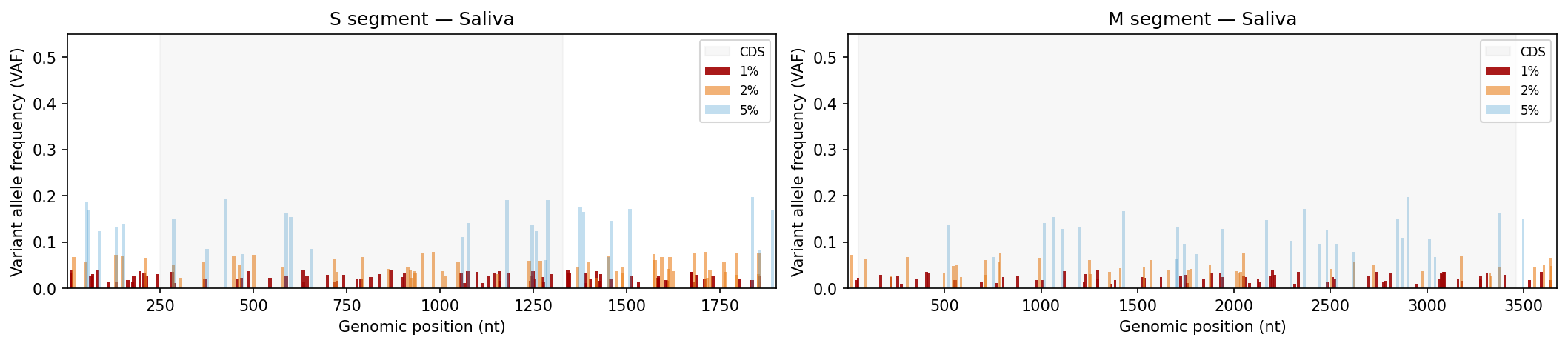
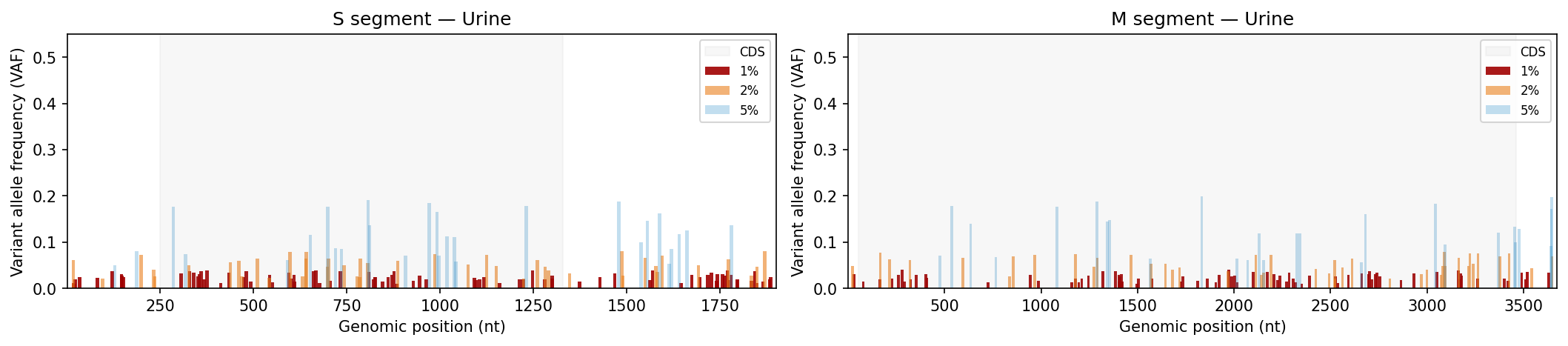
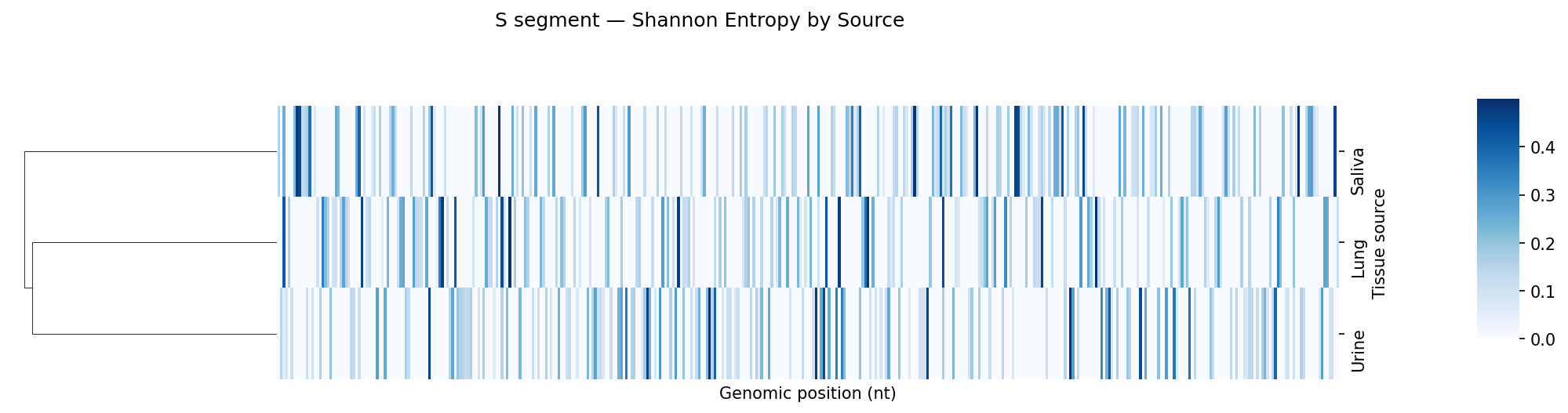
Mutation frequency, Shannon entropy heatmaps, and tissue-type comparisons
genomics
virology
VCF
mutation
entropy
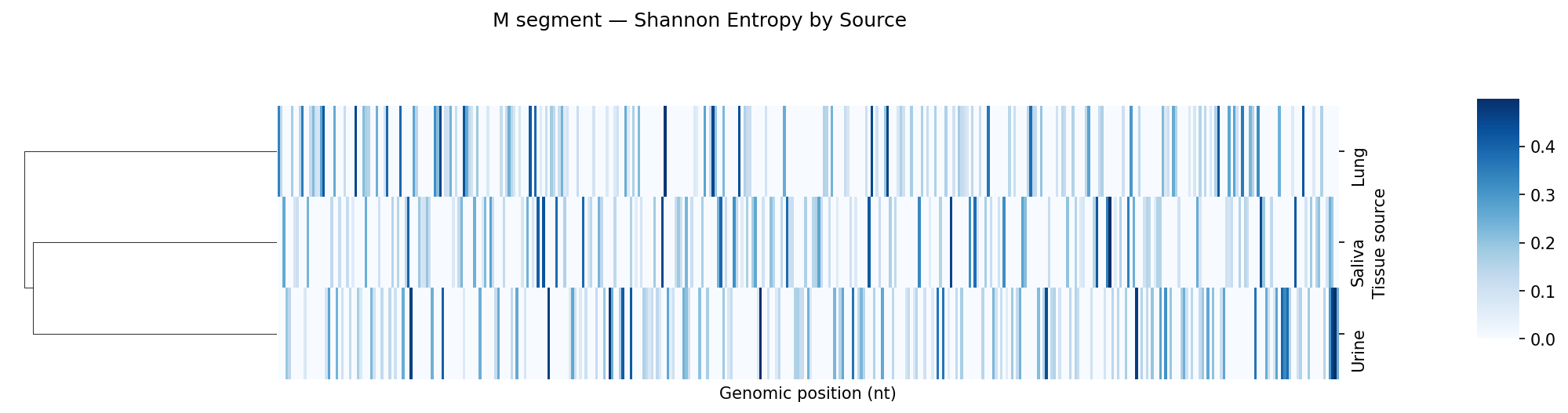
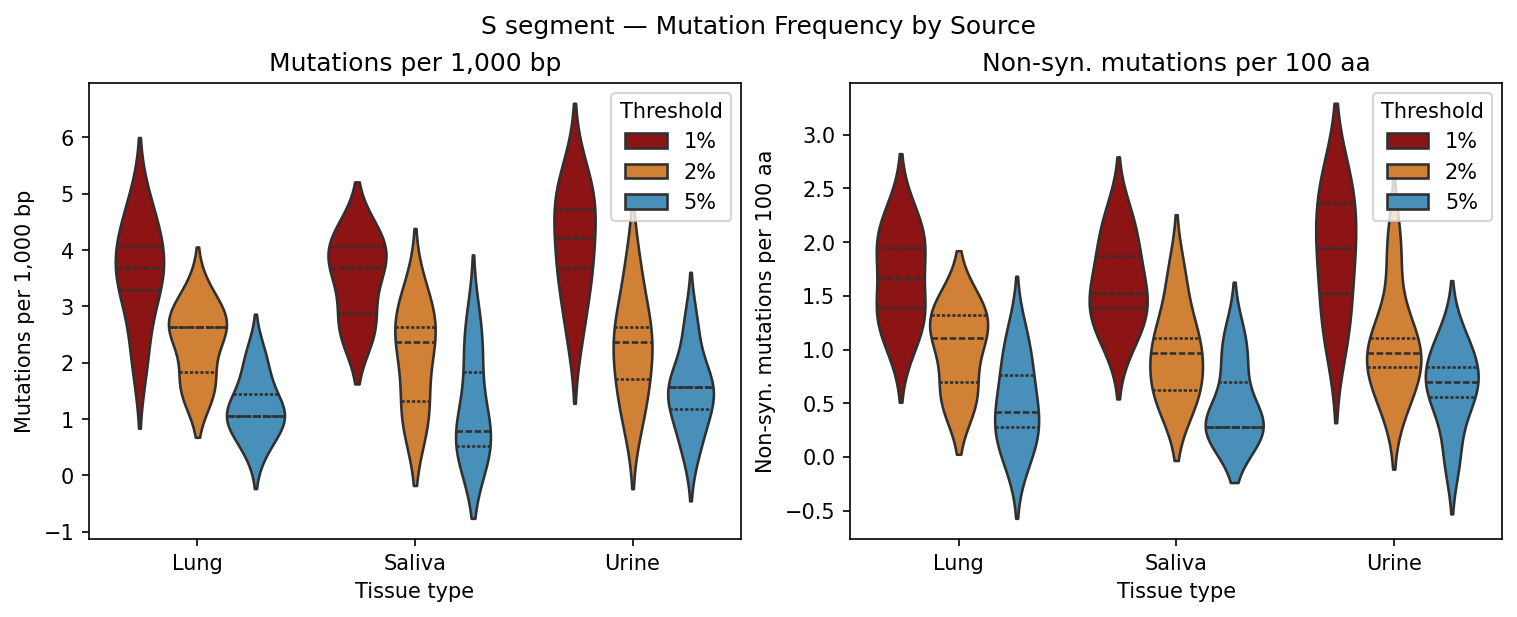
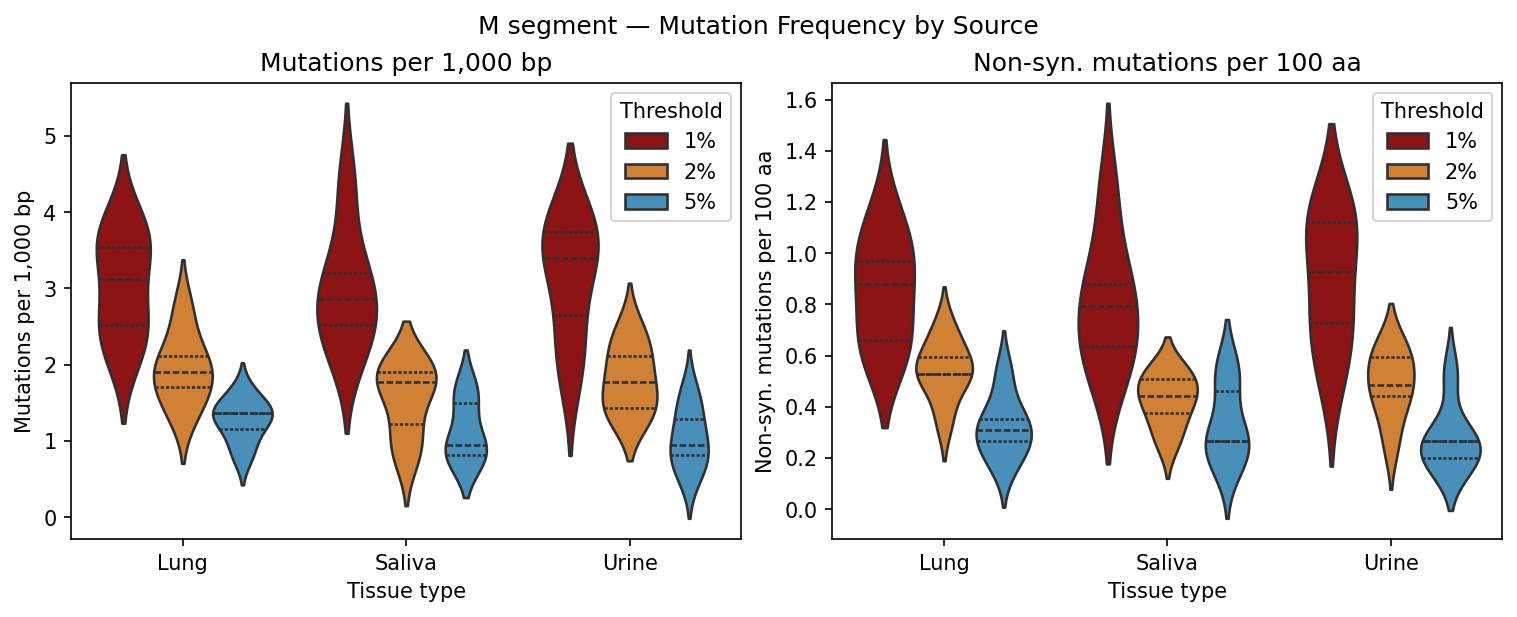
Per-sample VCF files from a variant caller. Coverage filtering, Shannon entropy per position, mutation frequency bar charts and entropy heatmaps per tissue type, Kruskal-Wallis comparison across tissue sources.
Overview
Item
Details
Input
Directory of .vcf files: aa_change_{ID}-{Source}_{Segment}_{Threshold}.vcf
Edit SEGMENT_CONFIG to match your pathogen’s genome (total length, CDS boundaries, AA count). TISSUE_TYPES controls which sources are included in statistical comparisons.
# ── USER CONFIGURATION ────────────────────────────────────────────────────────# Directory containing VCF files (relative to this file)VCF_DIR ="data/aa_change_vcf"# VCF filename format (regex). Named groups: id, source, segment, threshold# Default: aa_change_{id}-{source}_{segment}_{threshold}.vcfVCF_FILENAME_PATTERN =r"aa_change_(?P<id>[^-]+)-(?P<source>[^_]+)_(?P<segment>[SM])_(?P<threshold>\d+pct)\.vcf"# Map threshold labels in filenames to display stringsTHRESHOLD_LABEL_MAP = {"5pct": "5%", "2pct": "2%", "1pct": "1%"}# Minimum read depth to retain a variant (coverage QC)DEPTH_THRESHOLD =500# Tissue / source types to include in the analysisTISSUE_TYPES = ["Lung", "Saliva", "Urine"]# Segment geometry — edit to match your pathogen's genomeSEGMENT_CONFIG = {"S": {"length": 1902,"cds_start": 250,"cds_end": 1329,"aa_length": 360, },"M": {"length": 3675,"cds_start": 52,"cds_end": 3461,"aa_length": 1135, },}# VAF thresholds to display in bar charts (ordered low → high)VAF_THRESHOLDS = ["1%", "2%", "5%"]# Colors and transparency for each VAF threshold in bar chartsVAF_COLORS = {"5%": "#3594cc","2%": "#ea801c","1%": "#a00000",}VAF_ALPHA = {"5%": 0.3, "2%": 0.6, "1%": 0.9}# Output directory for SVG plots (relative to this file)OUTPUT_DIR ="Plots"# ─────────────────────────────────────────────────────────────────────────────
Setup
Code
import osimport reimport warningsimport numpy as npimport pandas as pdimport vcfpyimport matplotlib.pyplot as pltimport seaborn as snsfrom scipy.stats import entropy, kruskalwarnings.filterwarnings("ignore")plt.rcParams.update({'axes.titlesize': 12,'axes.labelsize': 10,'xtick.labelsize': 10,'ytick.labelsize': 10,'legend.fontsize': 10,})pd.set_option('display.width', 1000)os.makedirs(OUTPUT_DIR, exist_ok=True)print(f"Output will be saved to: {os.path.abspath(OUTPUT_DIR)}")
Output will be saved to: /home/runner/work/lab-bioinfo-templates/lab-bioinfo-templates/templates/08_vcf-mutation-analysis/Plots
Load & Parse VCF Files
Code
pattern = re.compile(VCF_FILENAME_PATTERN)records = []vcf_files = [f for f in os.listdir(VCF_DIR) if f.endswith(".vcf")]print(f"Found {len(vcf_files)} VCF files in {VCF_DIR}")for fname in vcf_files: m = pattern.match(fname)ifnot m:print(f" Skipping (name mismatch): {fname}")continue sample_id = m.group("id") source = m.group("source") segment = m.group("segment") thresh_raw = m.group("threshold") threshold = THRESHOLD_LABEL_MAP.get(thresh_raw, thresh_raw) fpath = os.path.join(VCF_DIR, fname)try: reader = vcfpy.Reader.from_path(fpath)for rec in reader:ifnot rec.ALT:continue alt_allele = rec.ALT[0].value if rec.ALT else"." aa_change = rec.INFO.get("AA_CHANGE", "N/A")ifisinstance(aa_change, list): aa_change = aa_change[0] sample_call = ( rec.call_for_sample["SAMPLE"]if"SAMPLE"in rec.call_for_sampleelse rec.calls[0] ) ad = sample_call.data.get("AD", [0, 0]) dp = sample_call.data.get("DP", 0) records.append({"Position": rec.POS,"Reference_Allele": rec.REF,"Alternate_Allele": alt_allele,"Quality": rec.QUAL,"Amino_Acid_Change": aa_change,"Allelic_Depth_Ref": ad[0] iflen(ad) >0else0,"Allelic_Depth_Alt": ad[1] iflen(ad) >1else0,"Total_Depth": dp,"Source": source,"Segment": segment,"Variant_Threshold": threshold,"ID": sample_id, })exceptExceptionas e:print(f" Error reading {fname}: {type(e).__name__}: {e}")combined_df = pd.DataFrame(records)print(f"Total records parsed: {len(combined_df)}")if combined_df.empty:raiseRuntimeError(f"No VCF records were parsed from '{os.path.abspath(VCF_DIR)}'. "f"Check that simulate_data.py ran successfully and that files match "f"VCF_FILENAME_PATTERN. Files found: {vcf_files[:5]}" )combined_df.head()
Found 108 VCF files in data/aa_change_vcf
Total records parsed: 881