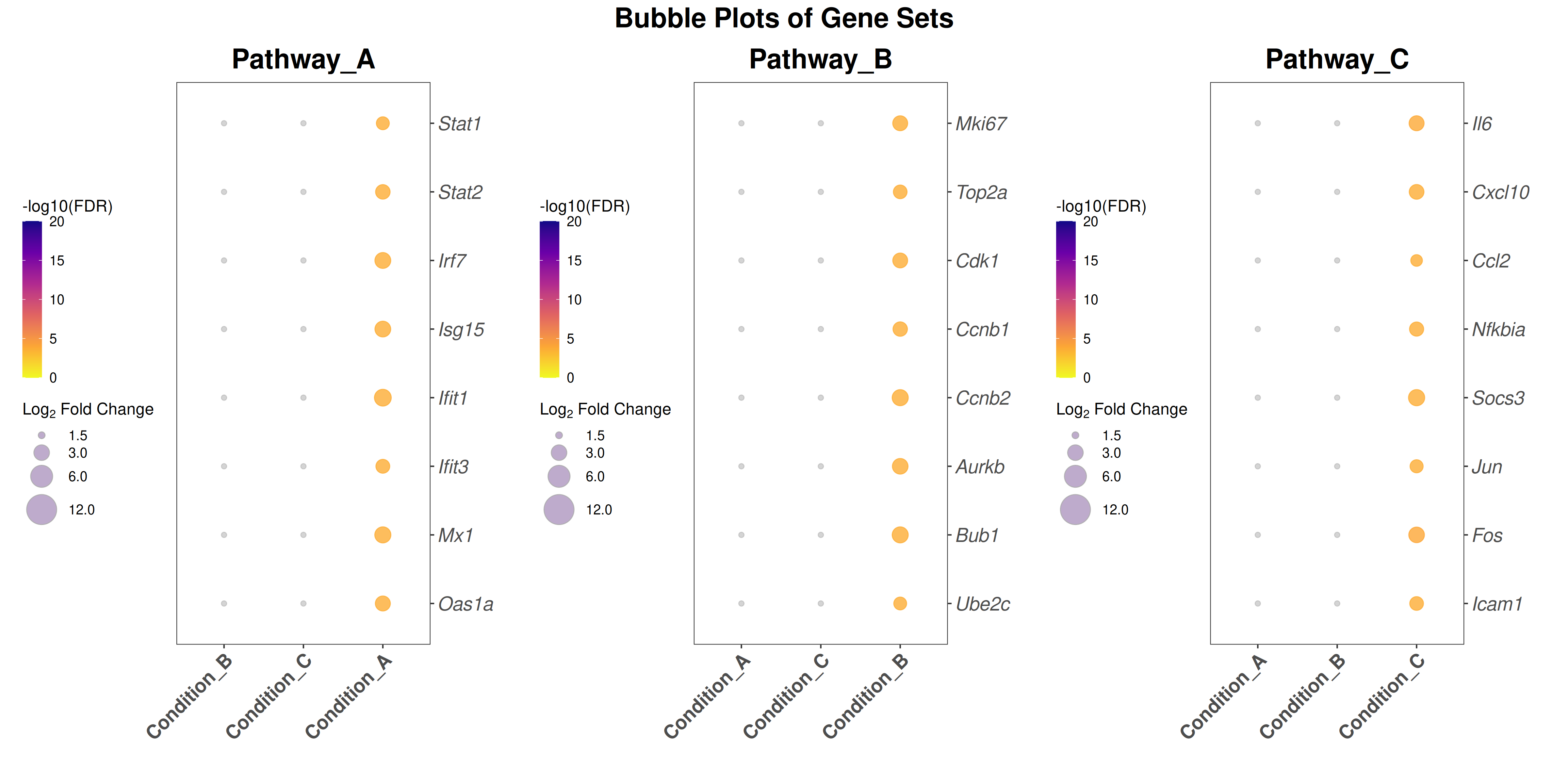
Gene Ontology, KEGG pathway enrichment, and multi-comparison bubble plots
genomics
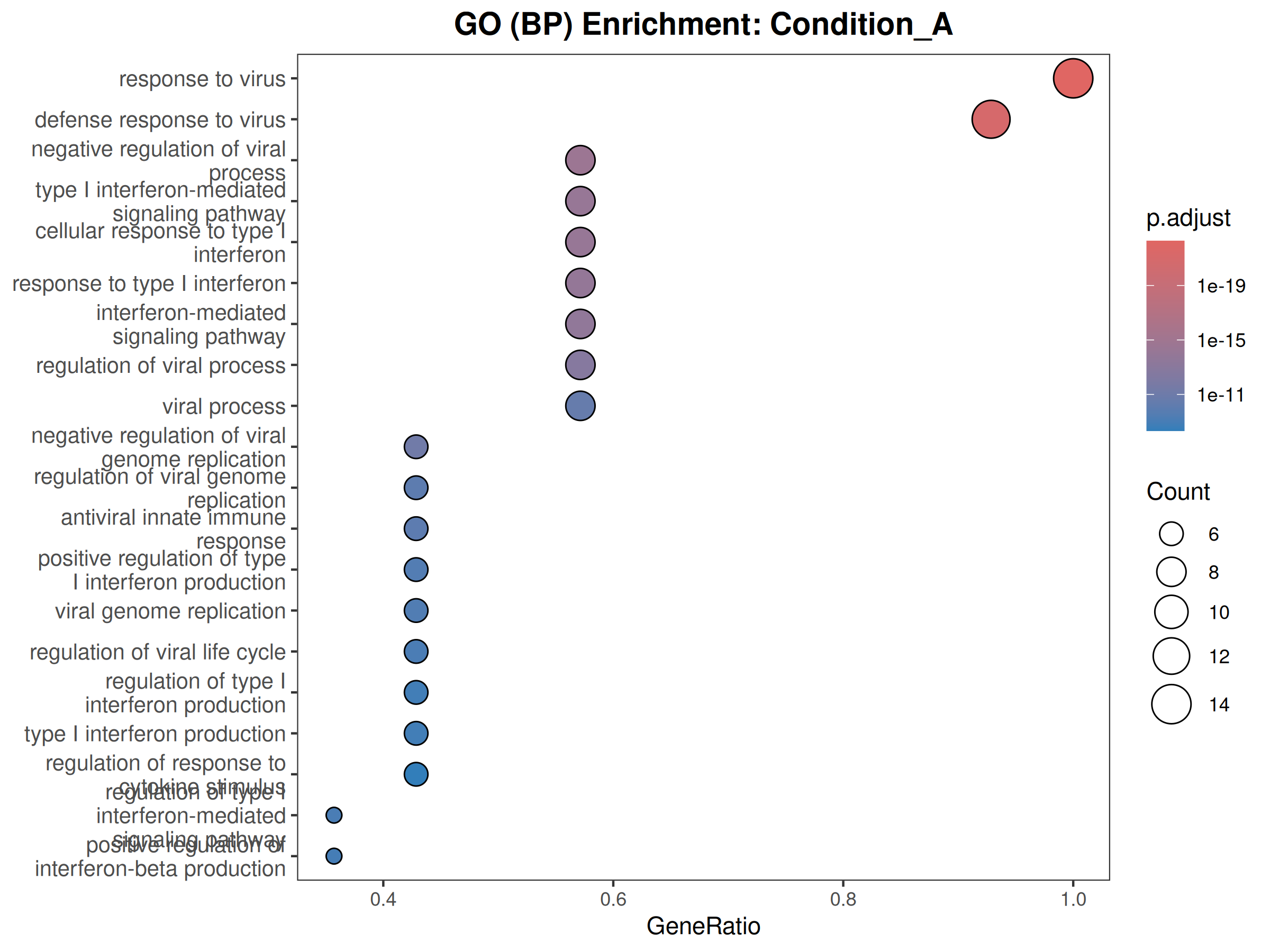
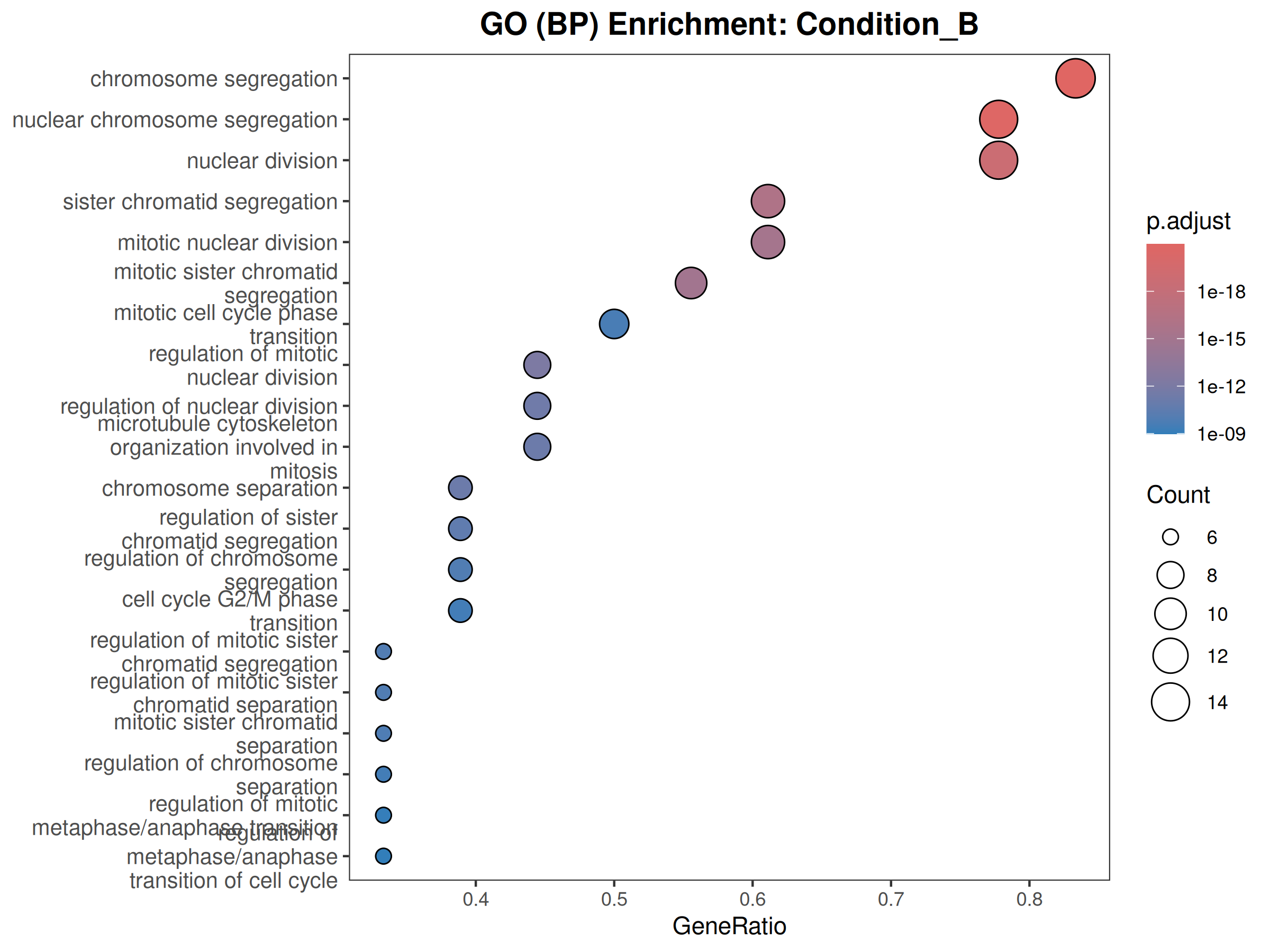
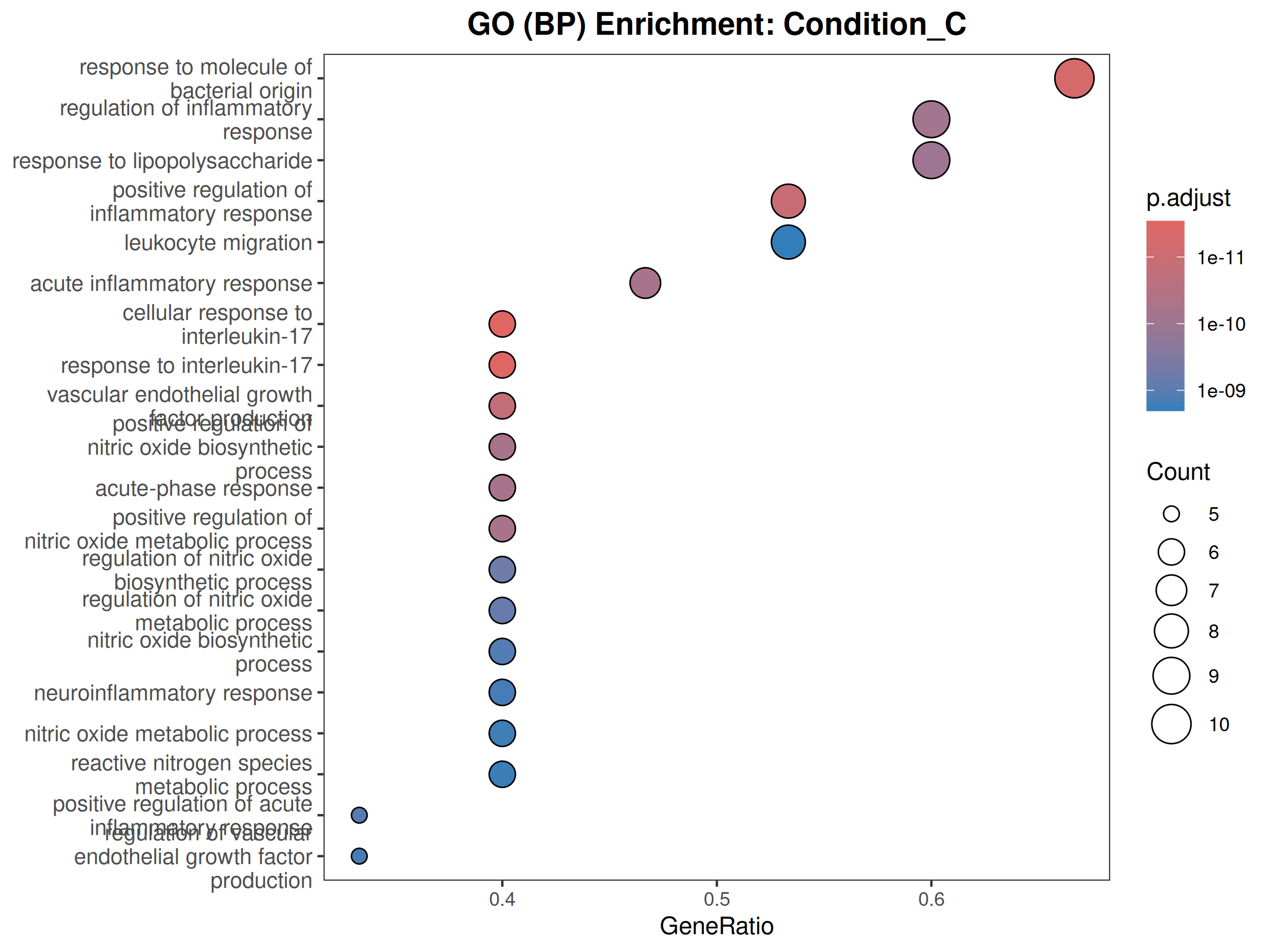
GO
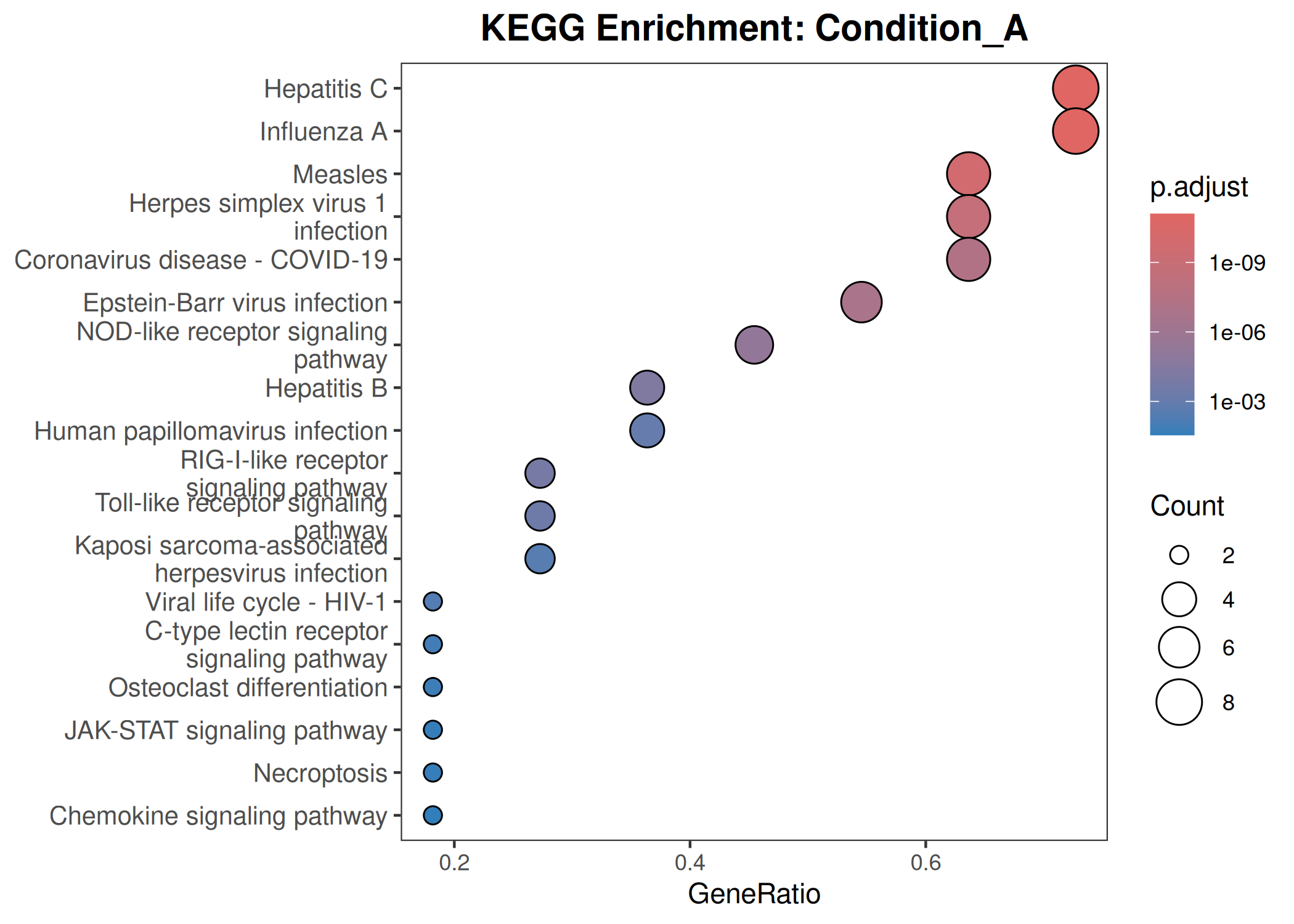
KEGG
enrichment
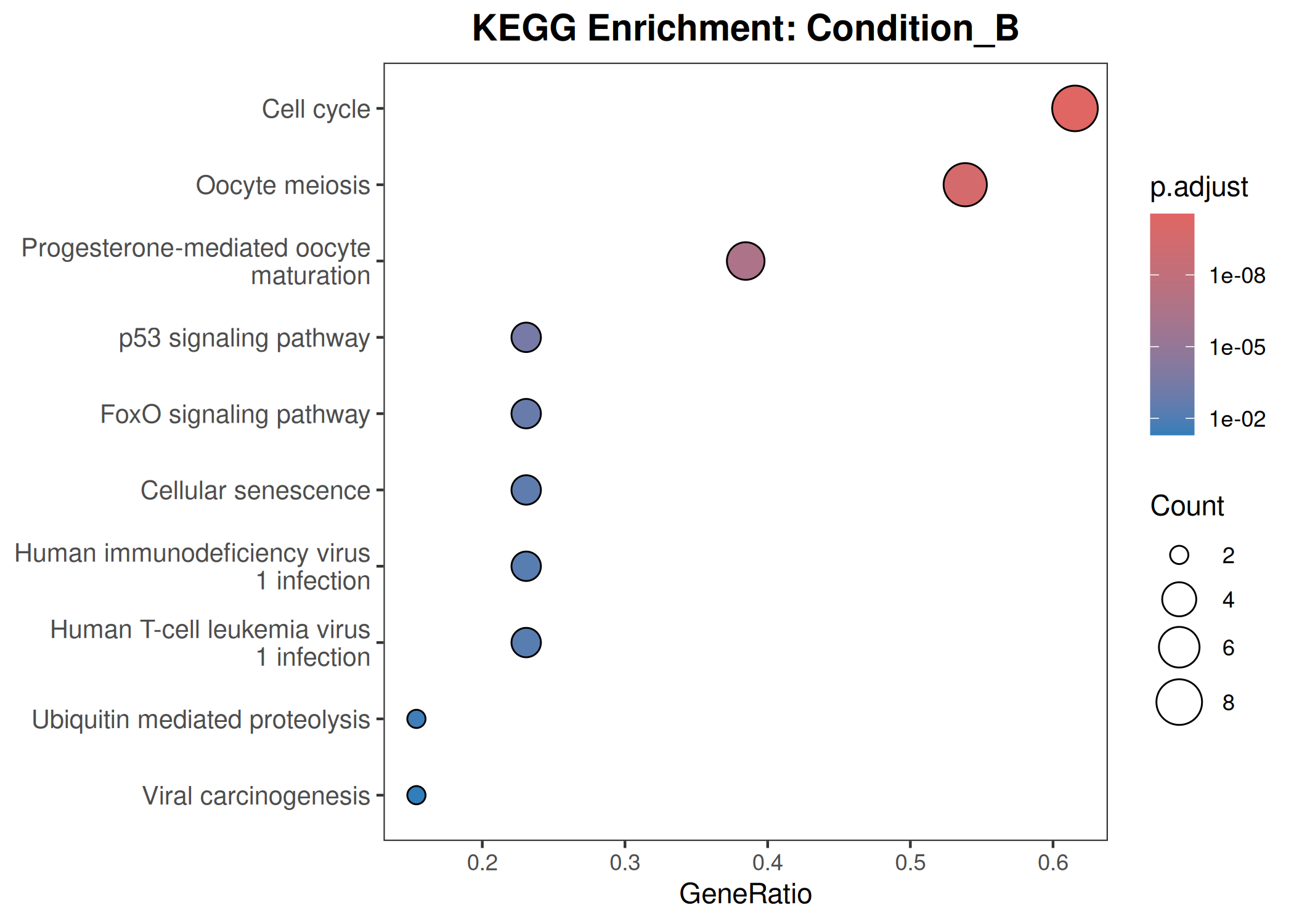
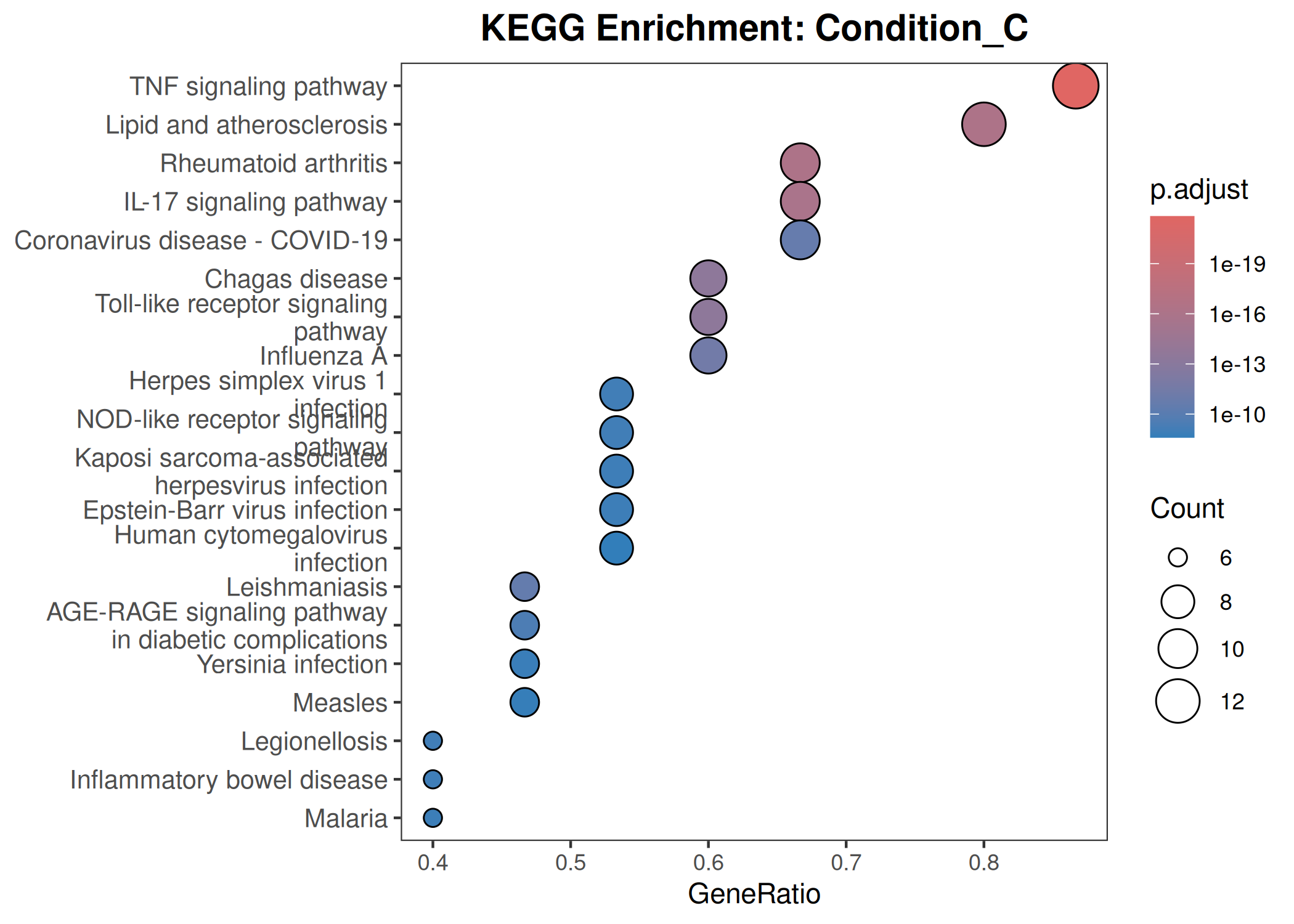
Performs GO (Biological Process) and KEGG pathway enrichment analysis across one or more DESeq2 comparisons. Produces publication-quality bubble plots for user-defined gene sets, enrichment dot plots, and wide-format CSV exports for GraphPad/Prism.
Overview
Item
Details
Input
CSV with columns: Gene, Comparison, log2FoldChange, padj
When to use this template: You have DESeq2 results from one or more comparisons and want to visualize the expression of curated gene sets as bubble plots, and run GO/KEGG enrichment on significant DEGs.
Note
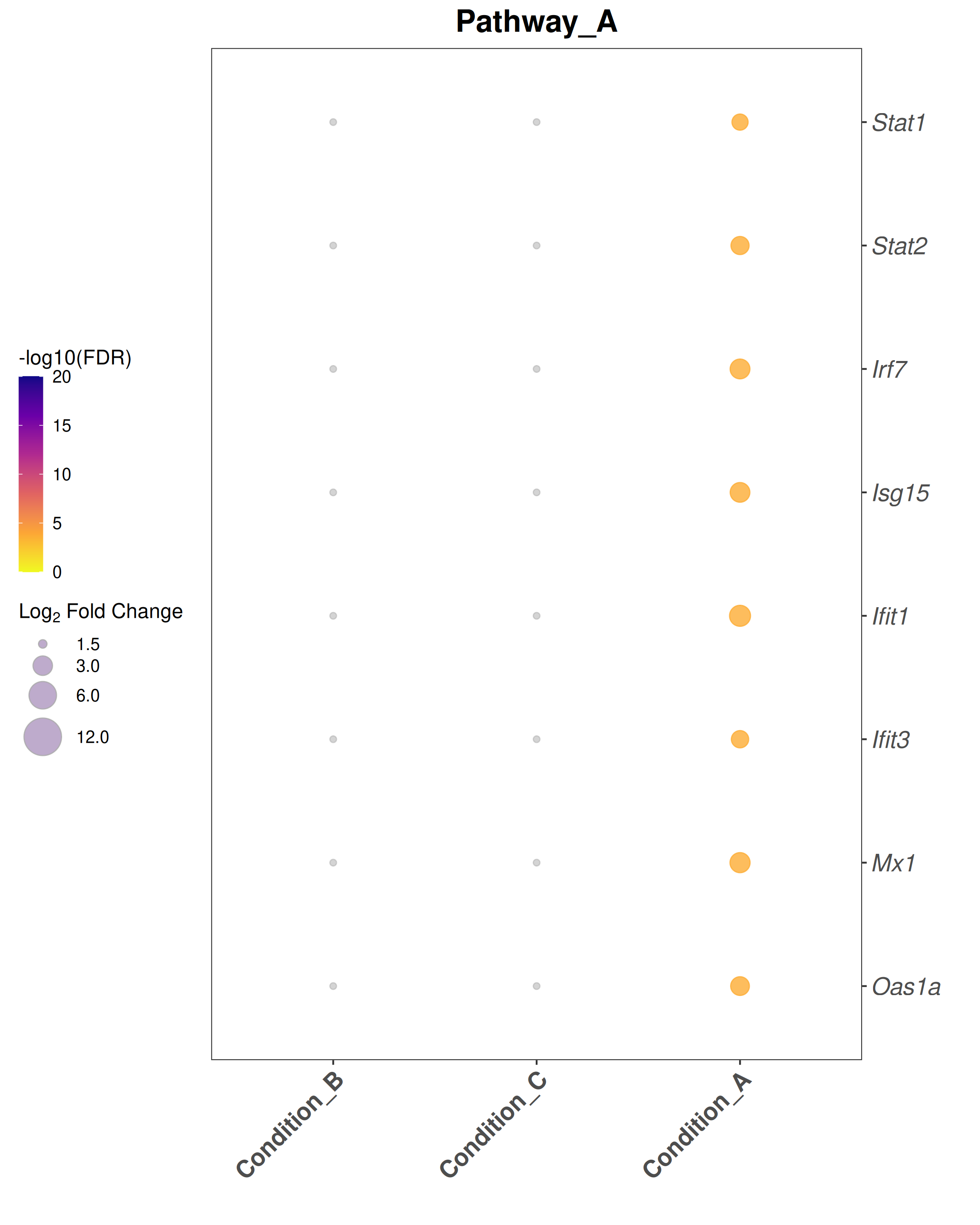
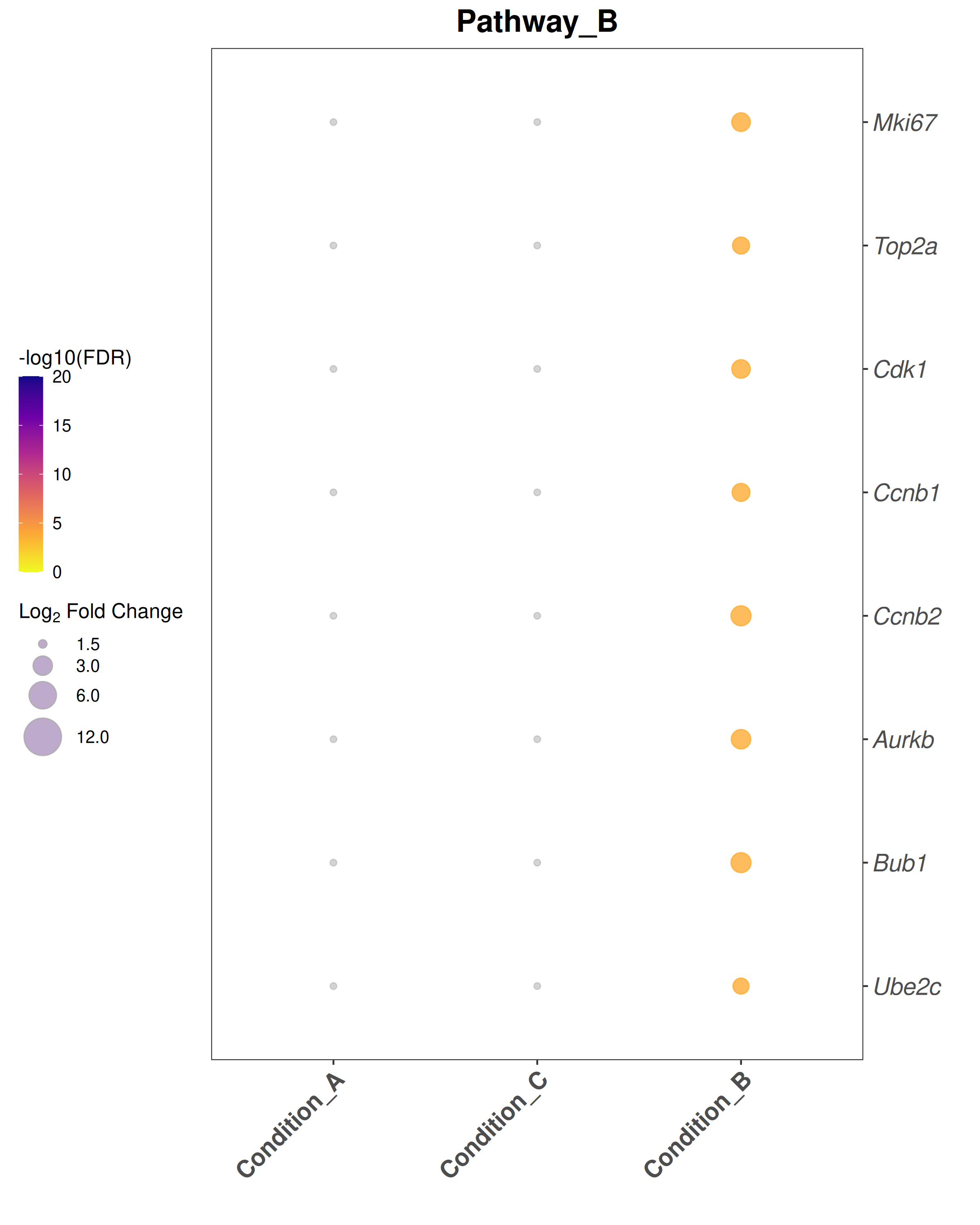
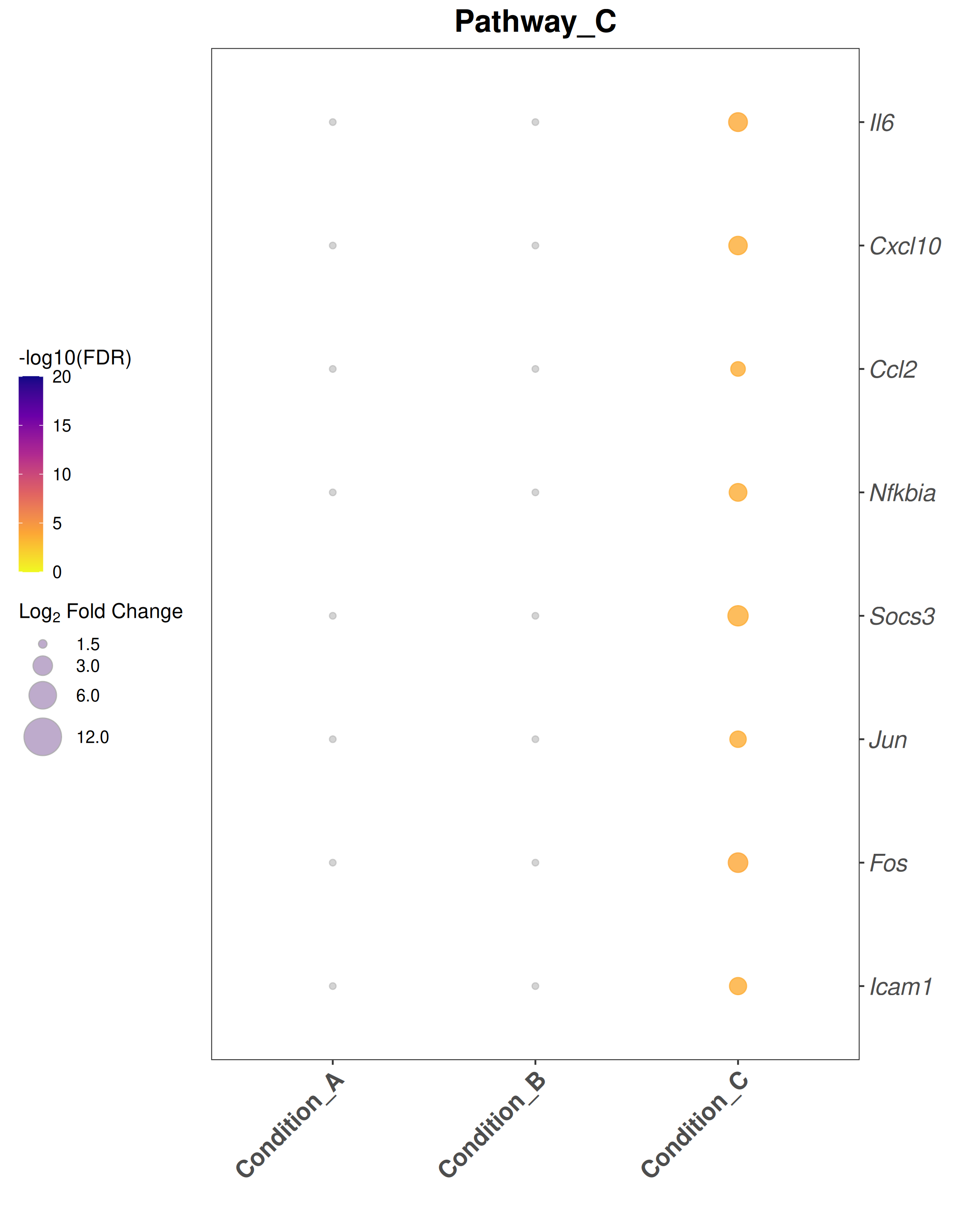
Bubble plot style:theme_bw() with blank panel grids · viridis plasma color scale · italic y-axis gene labels · legend on the left.
## ── USER CONFIGURATION ──────────────────────────────────────────────────────## DEG_FILE: CSV file with differential expression results.# Required columns: Gene, Comparison, log2FoldChange, padj# One row per gene per comparison.#DEG_FILE <-"data/deg_results.csv"# ORGANISM: Bioconductor annotation package for your species.# Human: "org.Hs.eg.db" Mouse: "org.Mm.eg.db"ORG_DB <-"org.Mm.eg.db"SPECIES <-"Mus musculus"# used in plot subtitles# FDR_THRESHOLD: Significance threshold for identifying DEGs.FDR_THRESHOLD <-0.05# LFC_THRESHOLD: |log2FoldChange| cutoff for bubble plots and enrichment.LFC_THRESHOLD <-1.5# ENRICHMENT_FDR: FDR cutoff for reporting enriched terms.ENRICHMENT_FDR <-0.05# MIN_GENE_SET / MAX_GENE_SET: Gene set size limits for enrichment.MIN_GENE_SET <-10MAX_GENE_SET <-500# COMPARISONS_ORDER: Factor order for the x-axis of bubble plots.# Must match the "Comparison" values in DEG_FILE.# NULL = use alphabetical order.COMPARISONS_ORDER <-NULL# e.g., c("Condition_A", "Condition_B", "Condition_C")# COMPARISON_LABELS: Human-readable x-axis labels (same length as COMPARISONS_ORDER).# NULL = use raw comparison names.COMPARISON_LABELS <-NULL# e.g., c("Group A vs Ctrl", "Group B vs Ctrl", ...)# GENE_SETS: Named list of character vectors for bubble-plot gene sets.# Each element is one panel / pathway.# Replace these with your gene lists of interest.GENE_SETS <-list(Pathway_A =c("Stat1", "Stat2", "Irf7", "Isg15", "Ifit1","Ifit3", "Mx1", "Oas1a"),Pathway_B =c("Mki67", "Top2a", "Cdk1", "Ccnb1", "Ccnb2","Aurkb", "Bub1", "Ube2c"),Pathway_C =c("Il6", "Cxcl10", "Ccl2", "Nfkbia", "Socs3","Jun", "Fos", "Icam1"))# OUTPUT_DIR: Where to save plots and data exports.OUTPUT_DIR <-"Plots"## ────────────────────────────────────────────────────────────────────────────