Viral titer quantification with replicate QC and significance testing
infection
virology
ANOVA
plaque-assay
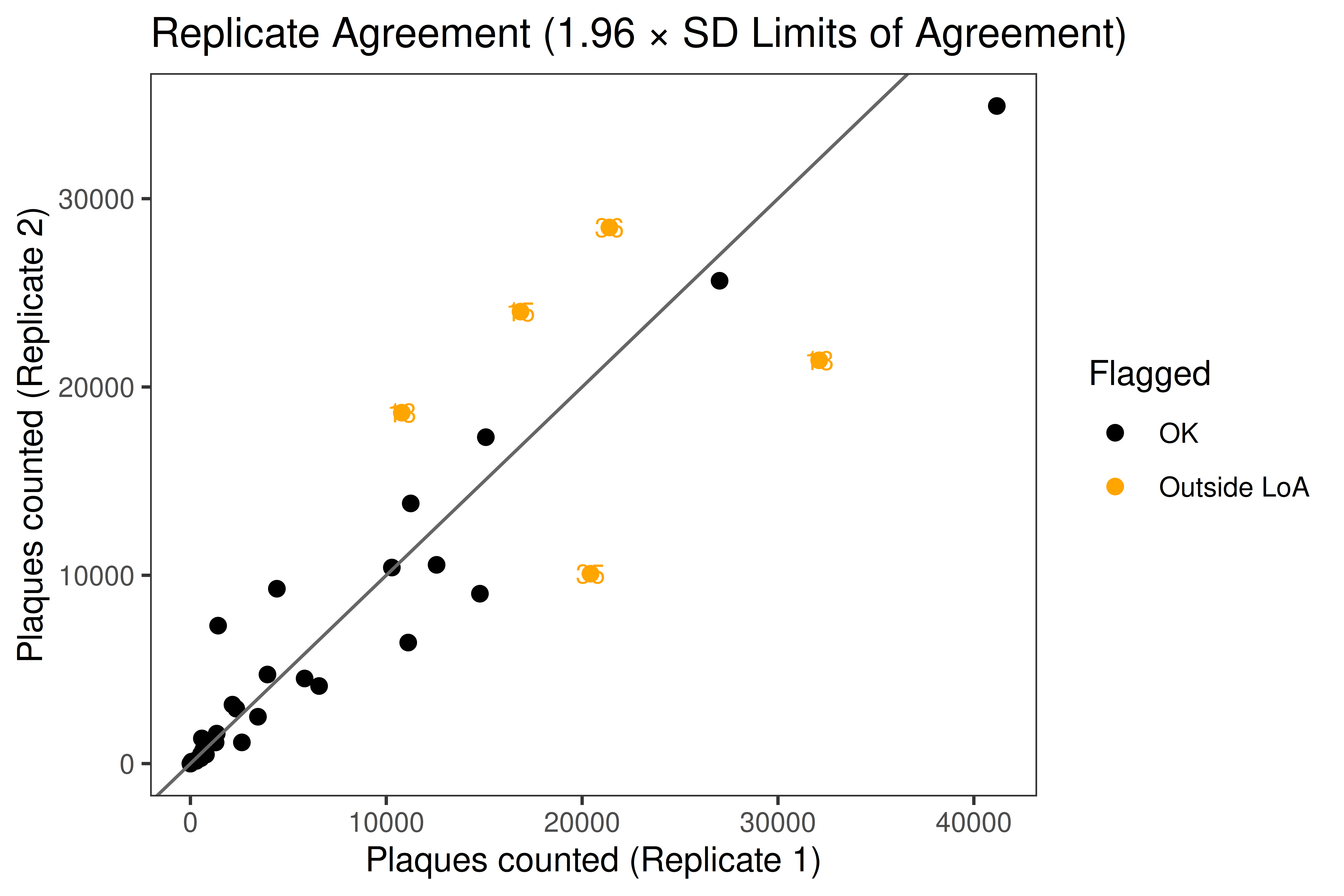
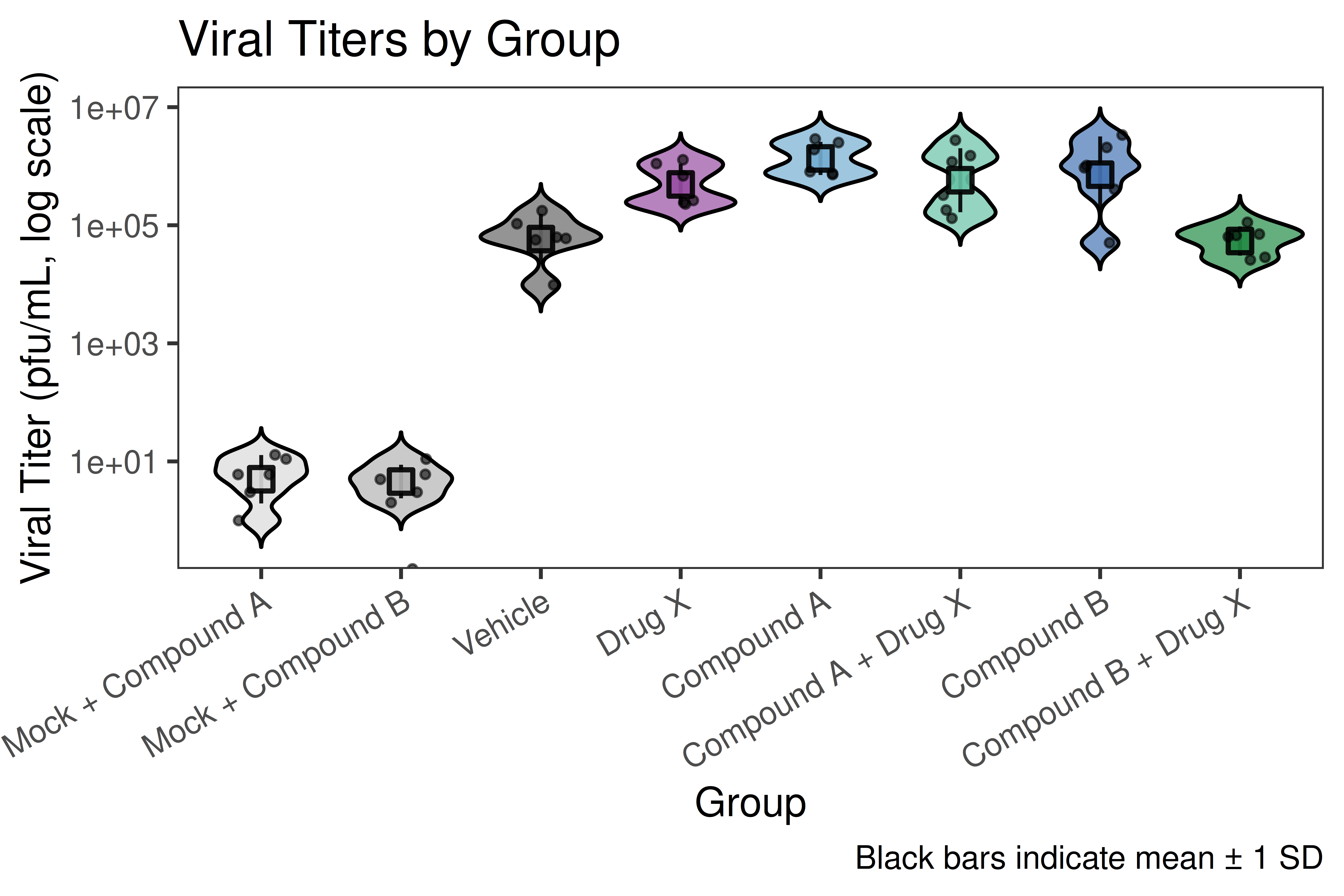
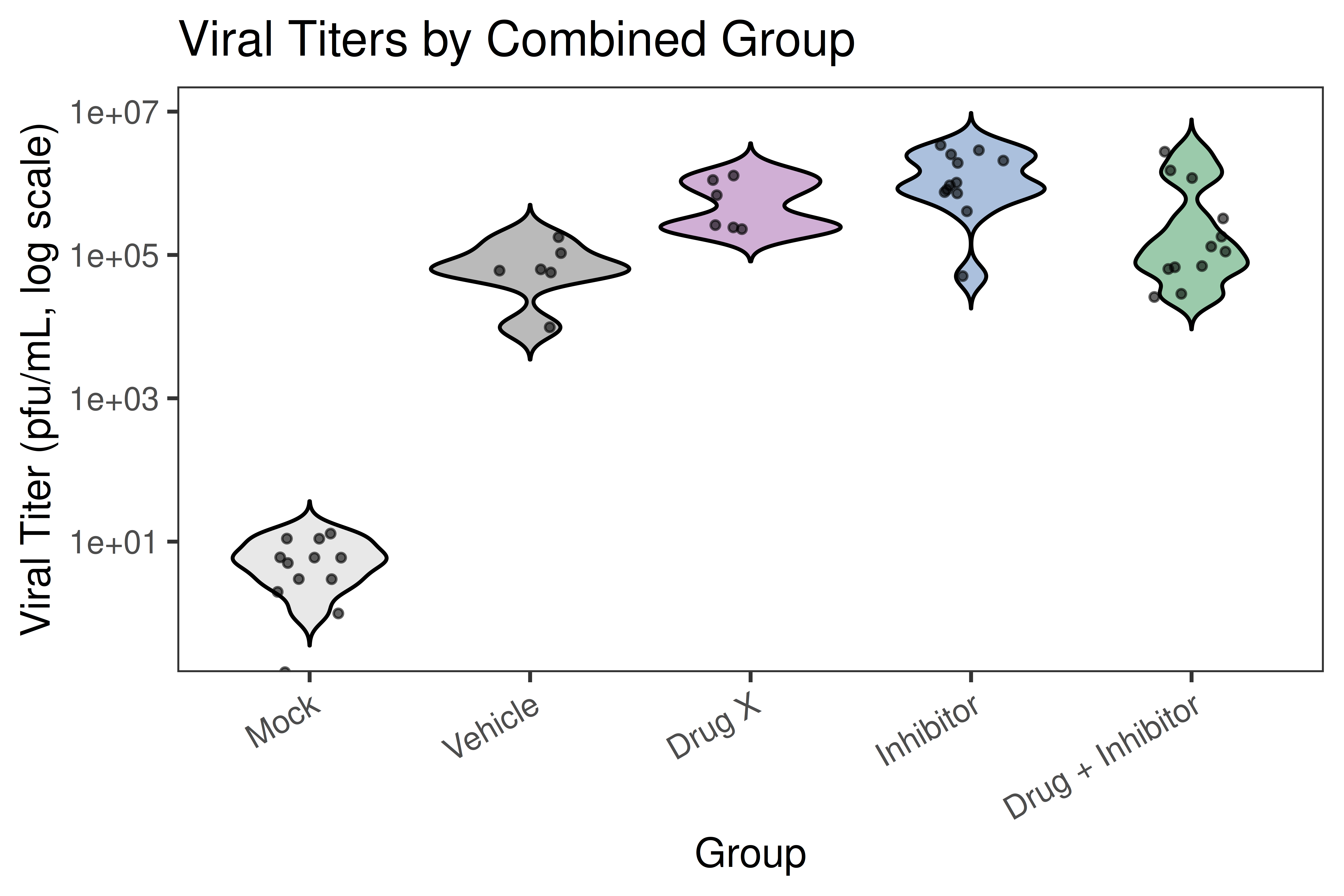
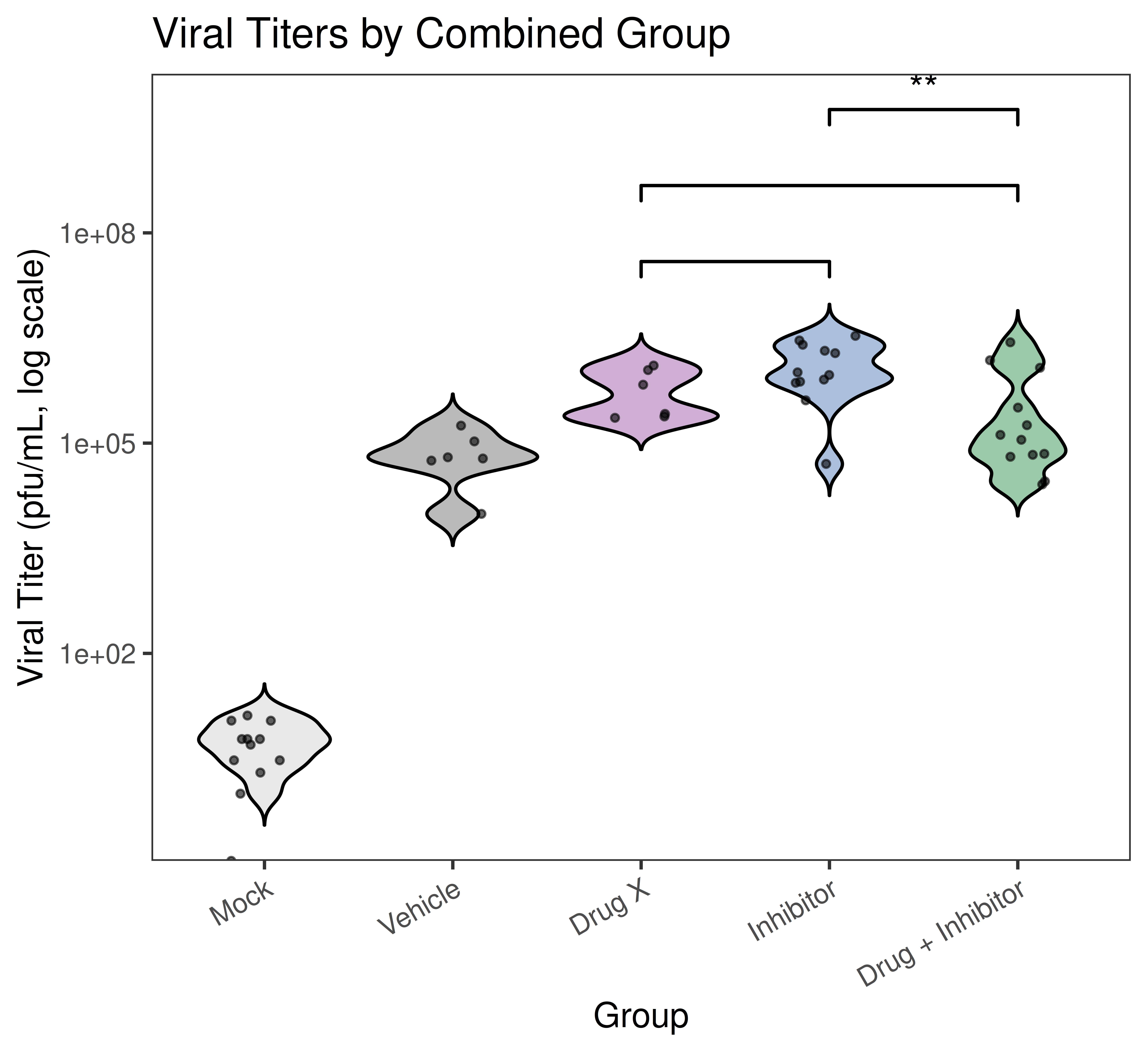
Analyzes plaque assay data from viral infection studies. Performs replicate agreement (Limits of Agreement), one-way ANOVA with Tukey HSD, and produces publication-quality violin plots on a log10 titer scale.
Overview
Item
Details
Input
Single CSV — one row per animal, columns for two plaque count replicates and calculated titer
When to use this template: You have viral plaque assay data with duplicate plaque counts per animal across multiple treatment groups, and you want to visualize titer distributions and test for significant differences.
## ── USER CONFIGURATION ──────────────────────────────────────────────────────## DATA_FILE: Path to your CSV file with plaque assay data.# Expected columns (rename yours to match, or update the rename() call below):# - "Mouse ID" : animal/sample identifier# - "Group" : group label (e.g., "Group 1", "Group 2")# - "Virus" : virus or mock label used in sample# - "# plaques counted R1" : plaque count replicate 1# - "# plaques counted R2" : plaque count replicate 2# - "Average plaques from duplicate wells in a 12-well plate"# - "Plated 10e-1 to 10e-6 dilution"# - "Total Volume (ml)"# - "pfu/mL right-side lung homogenate" : calculated viral titer#DATA_FILE <-"data/titer_data.csv"# EXCLUDE_PATTERN: Regex to drop rows you do not want (e.g., backtitrations).# Set to NULL to keep all rows.EXCLUDE_PATTERN <-NULL# e.g., "backtitration"# GROUP_MAP: Named character vector mapping your "Group N" labels to# human-readable short names used in all plots and tables.# Keys = values in the "Group" column of your CSV.# Values = display names (shown on plot axes).GROUP_MAP <-c("Group 1"="Mock + Compound A","Group 2"="Mock + Compound B","Group 3"="Compound A","Group 4"="Compound B","Group 5"="Drug X","Group 6"="Compound A + Drug X","Group 7"="Compound B + Drug X","Group 8"="Vehicle")# GROUP_ORDER: Display order on the x-axis. Must contain the same strings# as the values of GROUP_MAP.GROUP_ORDER <-c("Mock + Compound A","Mock + Compound B","Vehicle","Drug X","Compound A","Compound A + Drug X","Compound B","Compound B + Drug X")# PALETTE_GROUPS: Fill color for each group in the full violin plot.# Names must match the values of GROUP_MAP / GROUP_ORDER.PALETTE_GROUPS <-c("Mock + Compound A"="grey85","Mock + Compound B"="grey70","Vehicle"="grey40","Drug X"="#984ea3","Compound A"="#74add1","Compound B"="#4575b4","Compound A + Drug X"="#66c2a5","Compound B + Drug X"="#238b45")# MERGE_MAP: Optionally collapse related groups for the combined-group plot.# Keys = ShortGroup values; Values = merged display name.# Groups not listed here are kept as-is.MERGE_MAP <-c("Mock + Compound A"="Mock","Mock + Compound B"="Mock","Compound A"="Inhibitor","Compound B"="Inhibitor","Compound A + Drug X"="Drug + Inhibitor","Compound B + Drug X"="Drug + Inhibitor","Vehicle"="Vehicle","Drug X"="Drug X")# MERGE_ORDER: Display order for the combined-group violin plot.MERGE_ORDER <-c("Mock", "Vehicle", "Drug X", "Inhibitor", "Drug + Inhibitor")# PALETTE_MERGED: Fill colors for the combined-group violin plot.PALETTE_MERGED <-c("Mock"="grey80","Vehicle"="grey40","Drug X"="#984ea3","Inhibitor"="#4575b4","Drug + Inhibitor"="#238b45")# COMPARISONS: Pairs of merged groups to test with stat_compare_means().# Each element is a length-2 character vector of MERGE_ORDER values.COMPARISONS <-list(c("Drug X", "Inhibitor"),c("Drug X", "Drug + Inhibitor"),c("Inhibitor", "Drug + Inhibitor"))# TITER_COL: Column name in your CSV that holds the computed titer value.TITER_COL <-"pfu/mL right-side lung homogenate"## ────────────────────────────────────────────────────────────────────────────
---title: "01 · Plaque Assay + Violin Plots"subtitle: "Viral titer quantification with replicate QC and significance testing"description: | Analyzes plaque assay data from viral infection studies. Performs replicate agreement (Limits of Agreement), one-way ANOVA with Tukey HSD, and produces publication-quality violin plots on a log10 titer scale.categories: [infection, virology, ANOVA, plaque-assay]---## Overview| Item | Details ||------|---------|| **Input** | Single CSV — one row per animal, columns for two plaque count replicates and calculated titer || **Key packages** | `ggplot2`, `ggpubr`, `rstatix`, `car`, `gt`, `scales` || **Statistics** | Limits of Agreement · Shapiro-Wilk · Levene · One-way ANOVA · Tukey HSD || **Output** | Replicate QC plot · Violin plots (full + merged) · ANOVA + post-hoc tables || **Download** | [template.Rmd](template.Rmd) |::: {.callout-tip}**When to use this template:** You have viral plaque assay data with duplicate plaquecounts per animal across multiple treatment groups, and you want to visualize titerdistributions and test for significant differences.:::[Back to Gallery](../../index.html){.btn .btn-outline-secondary}[Open Template File](template.Rmd){.btn .btn-primary}```{r child="template.Rmd"}```