End-to-end differential expression from counts or SummarizedExperiment inputs
transcriptomics
RNA-seq
DESeq2
differential-expression
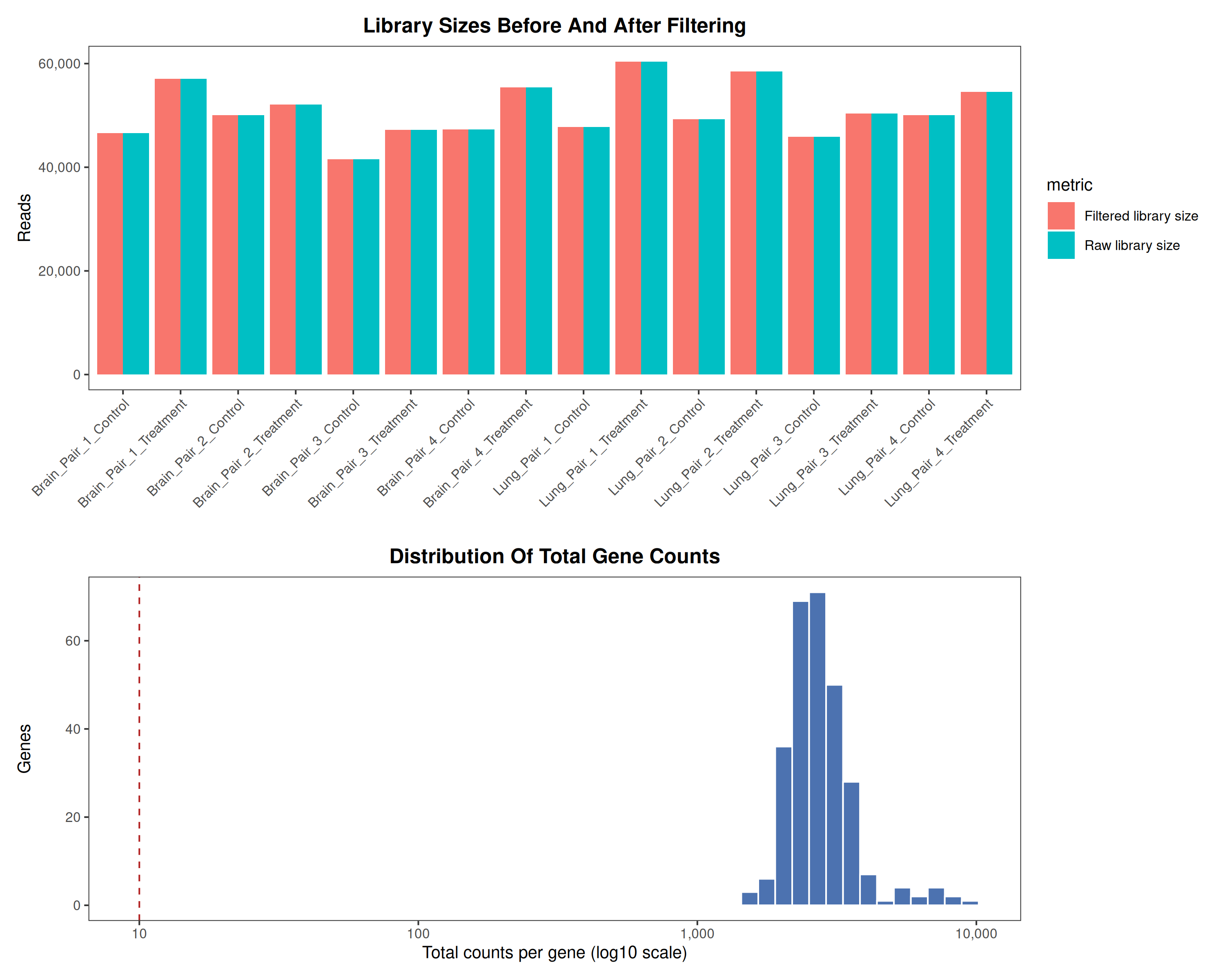
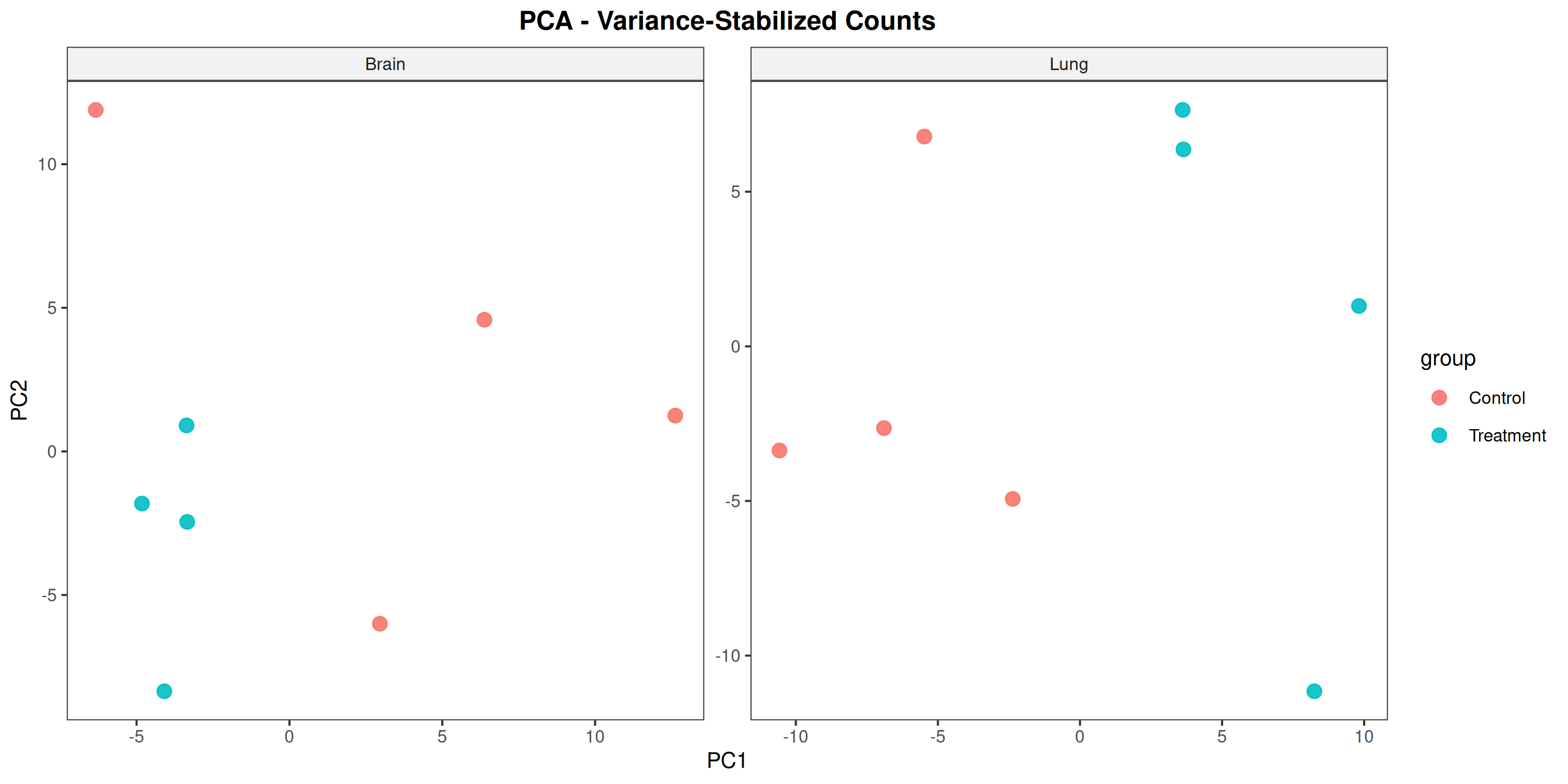
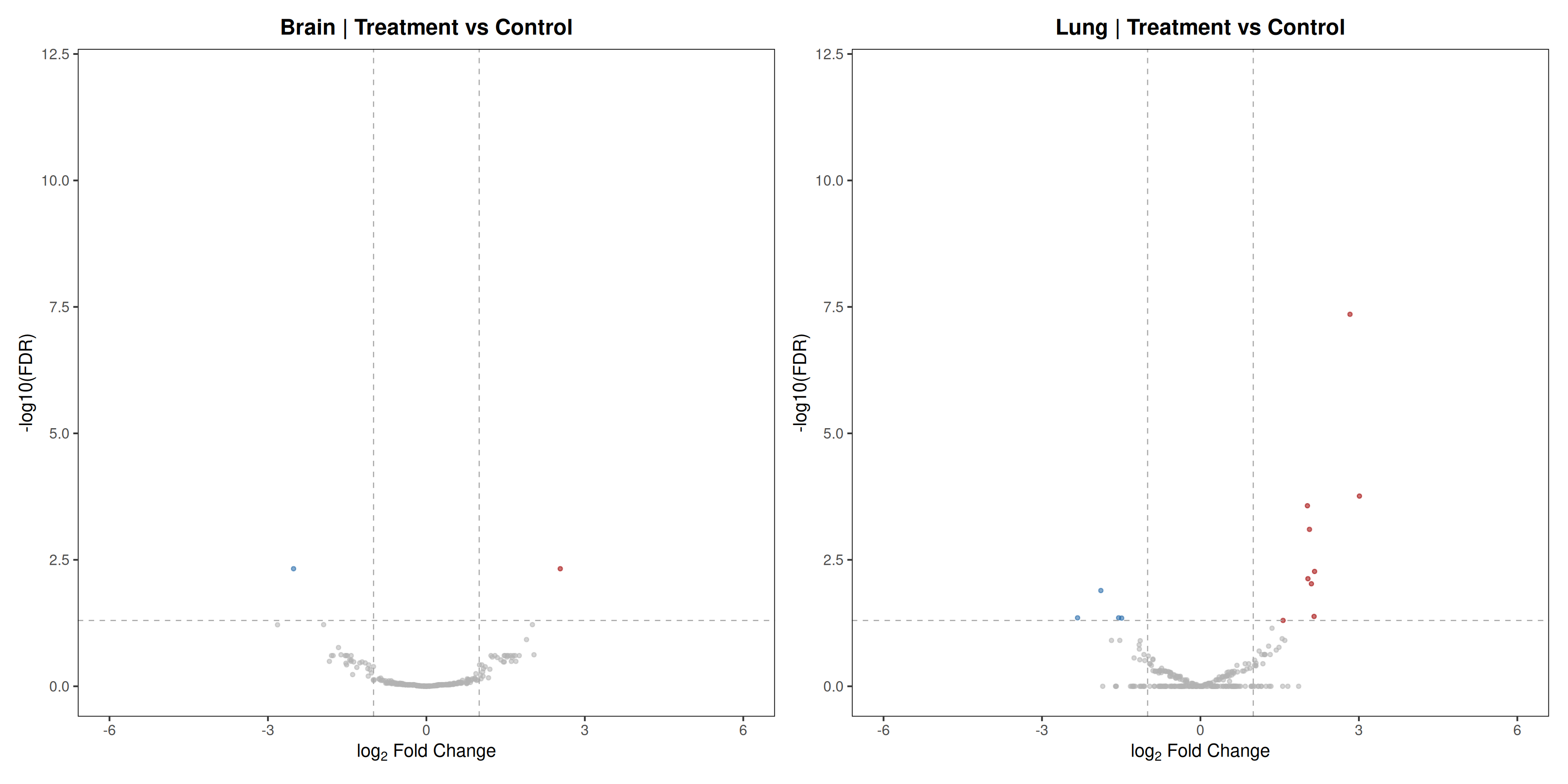
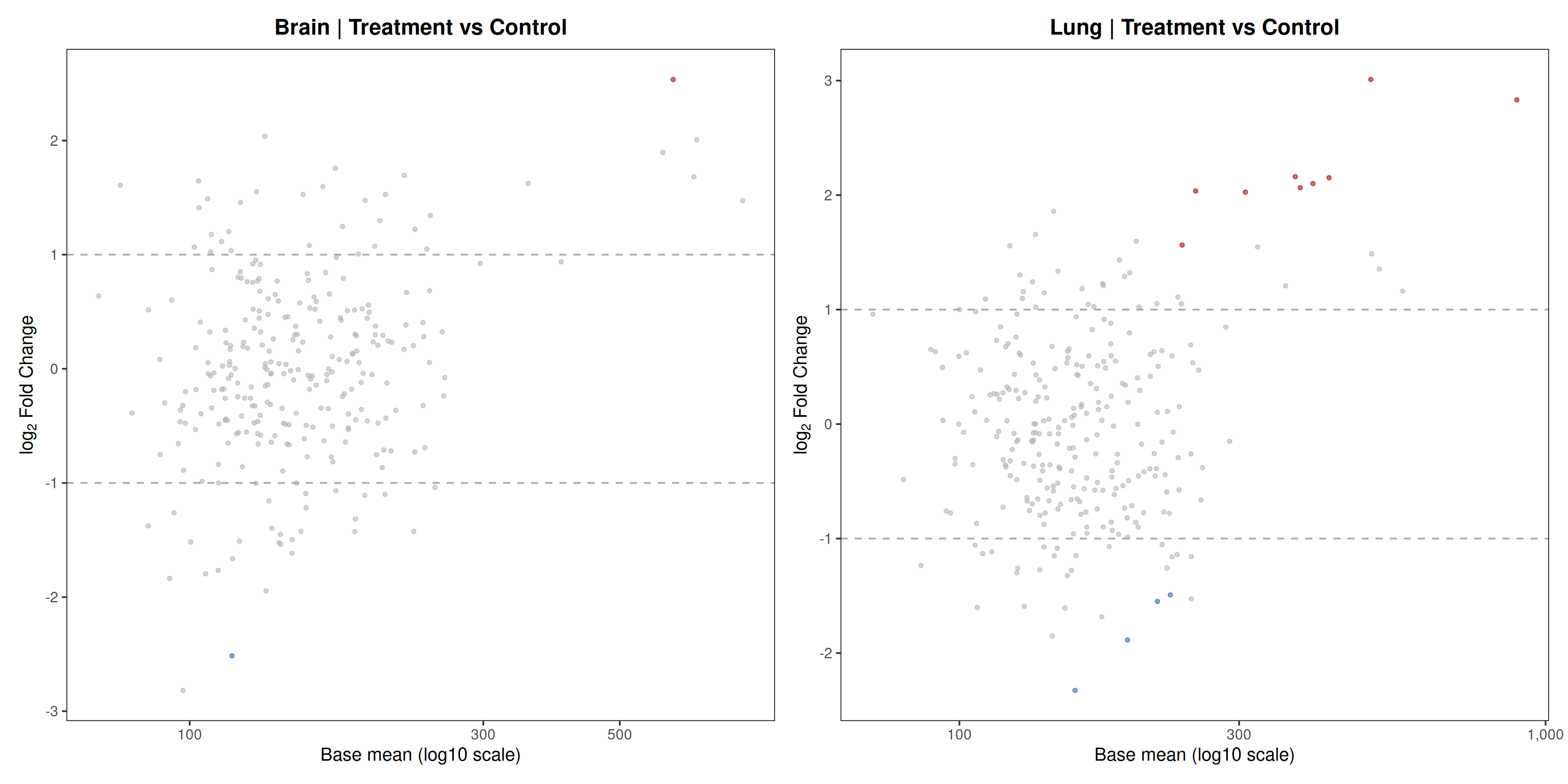
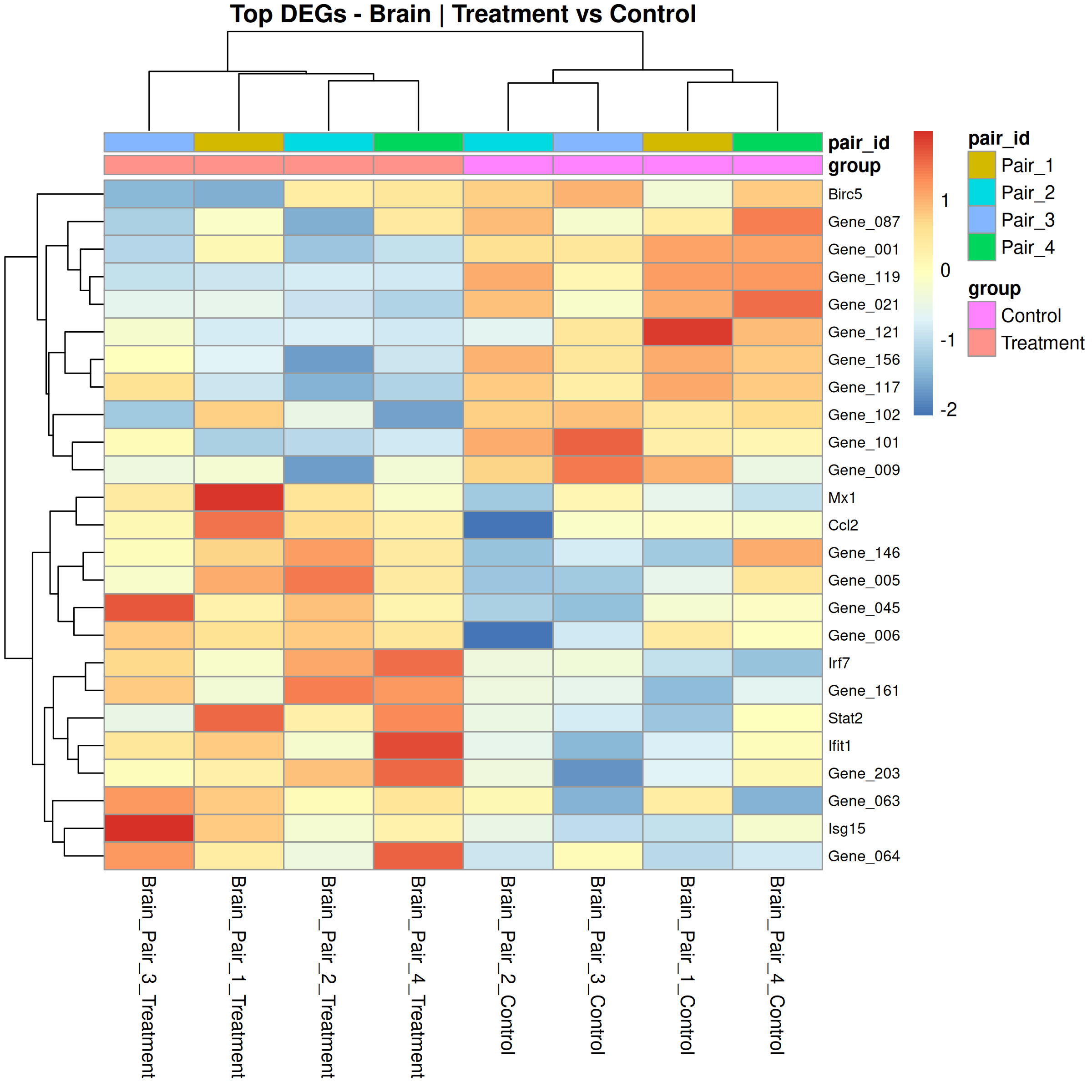
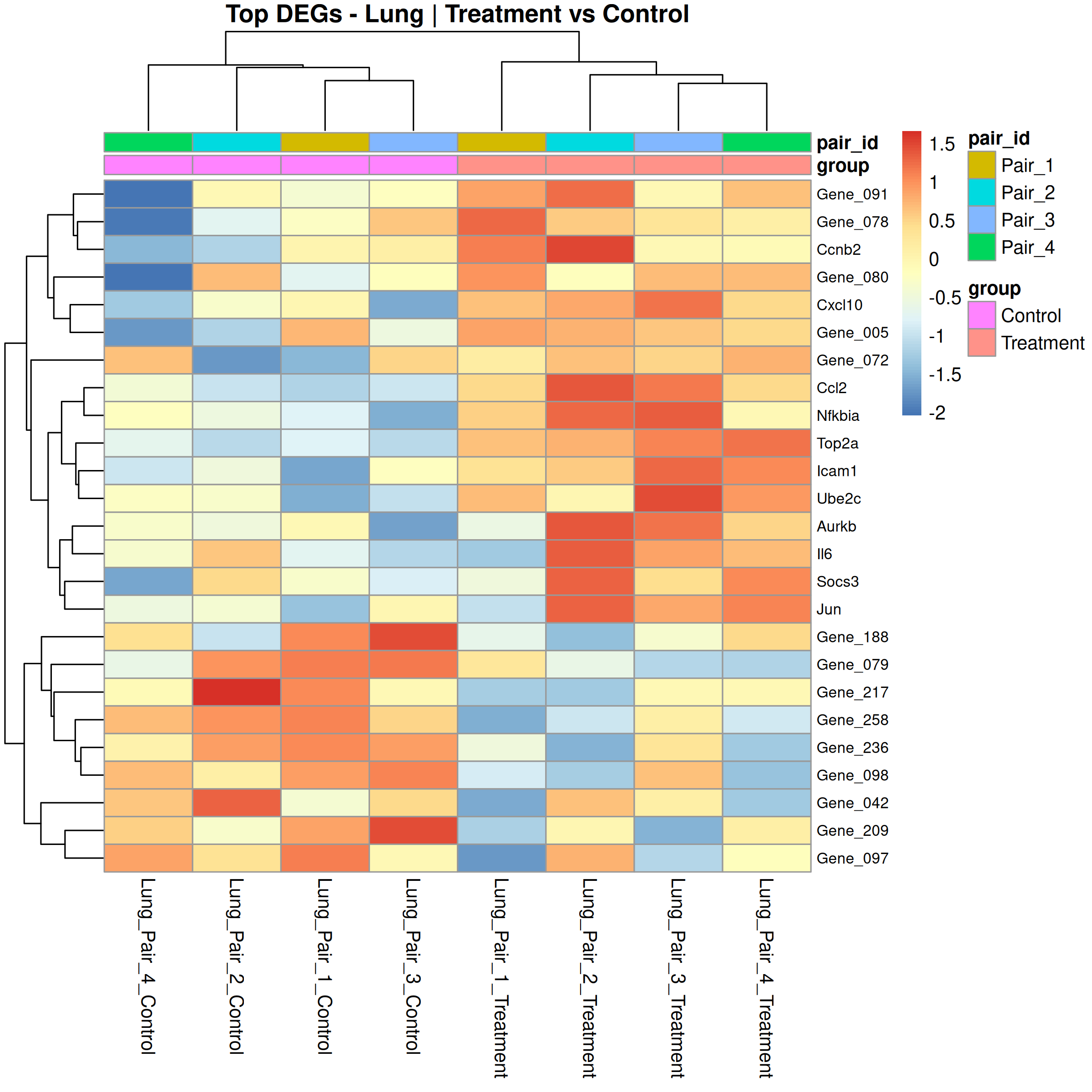
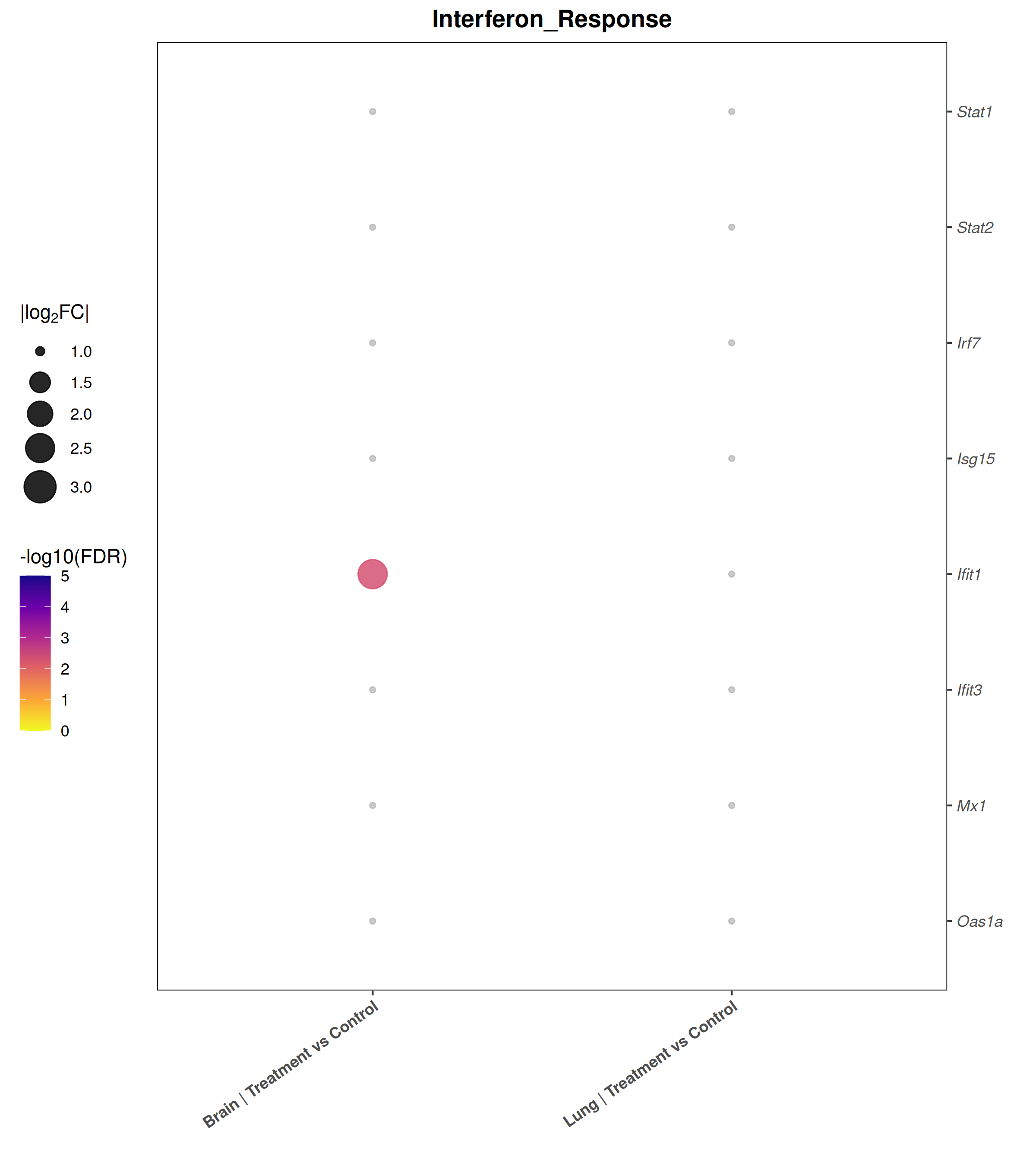
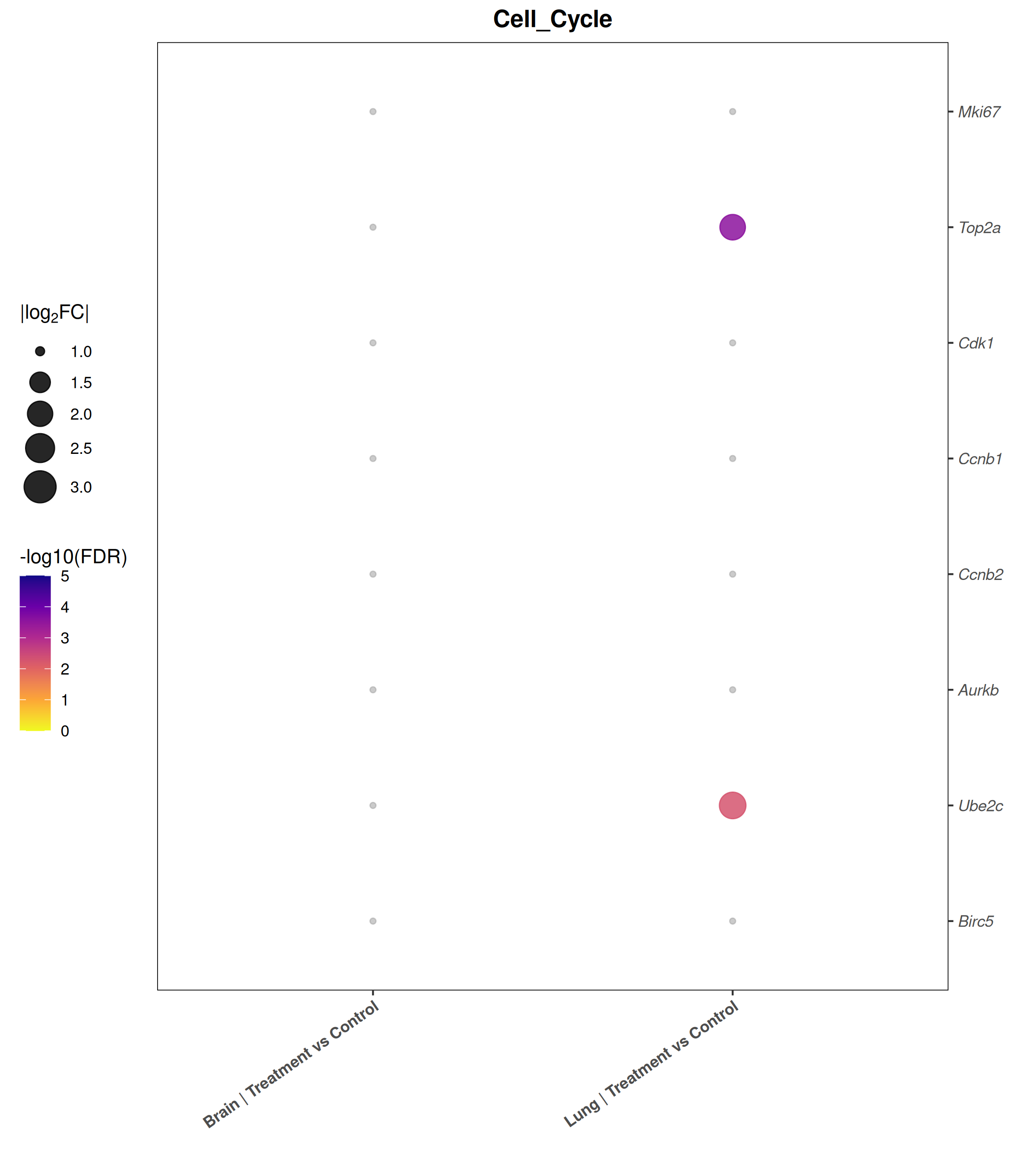
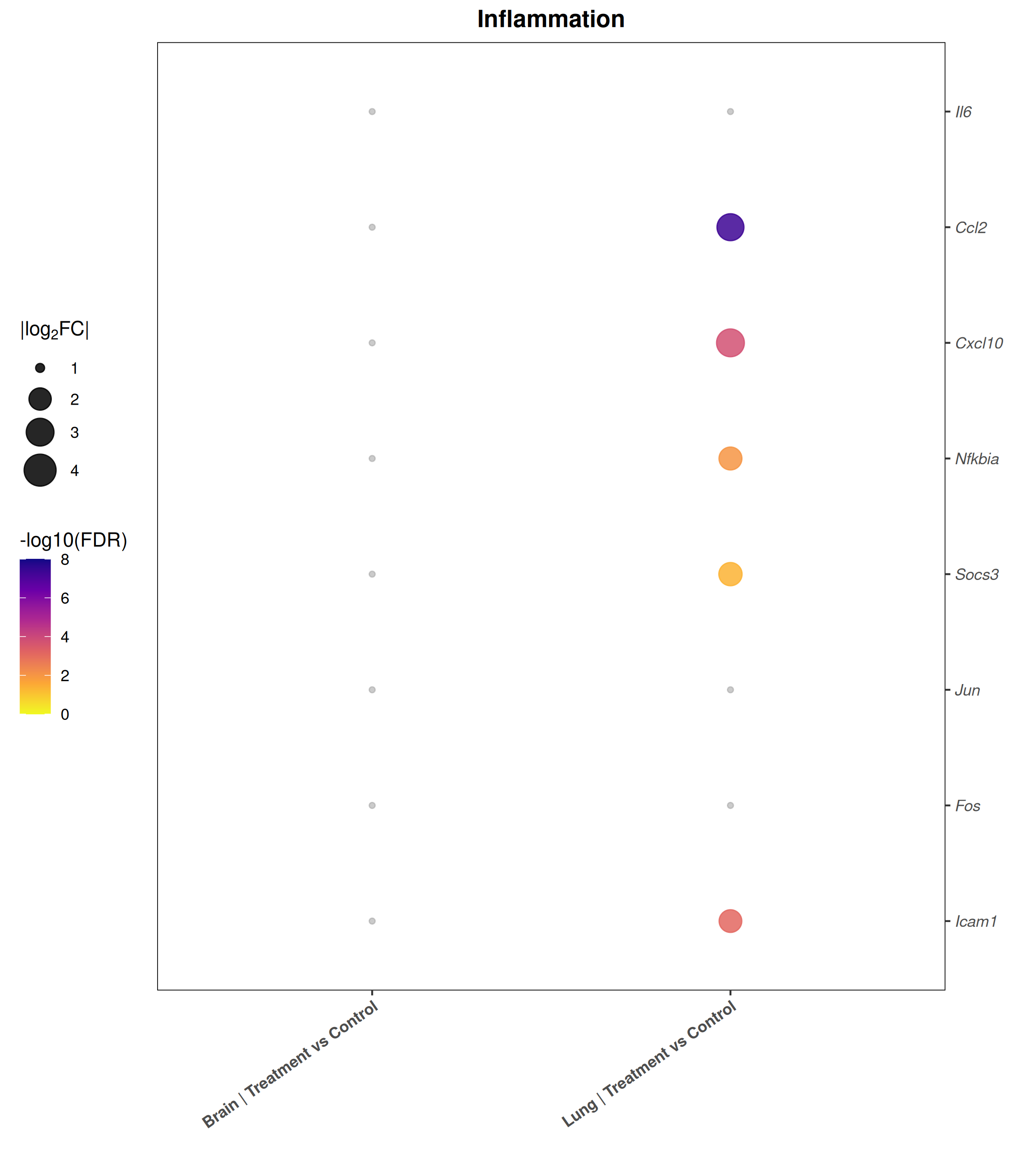
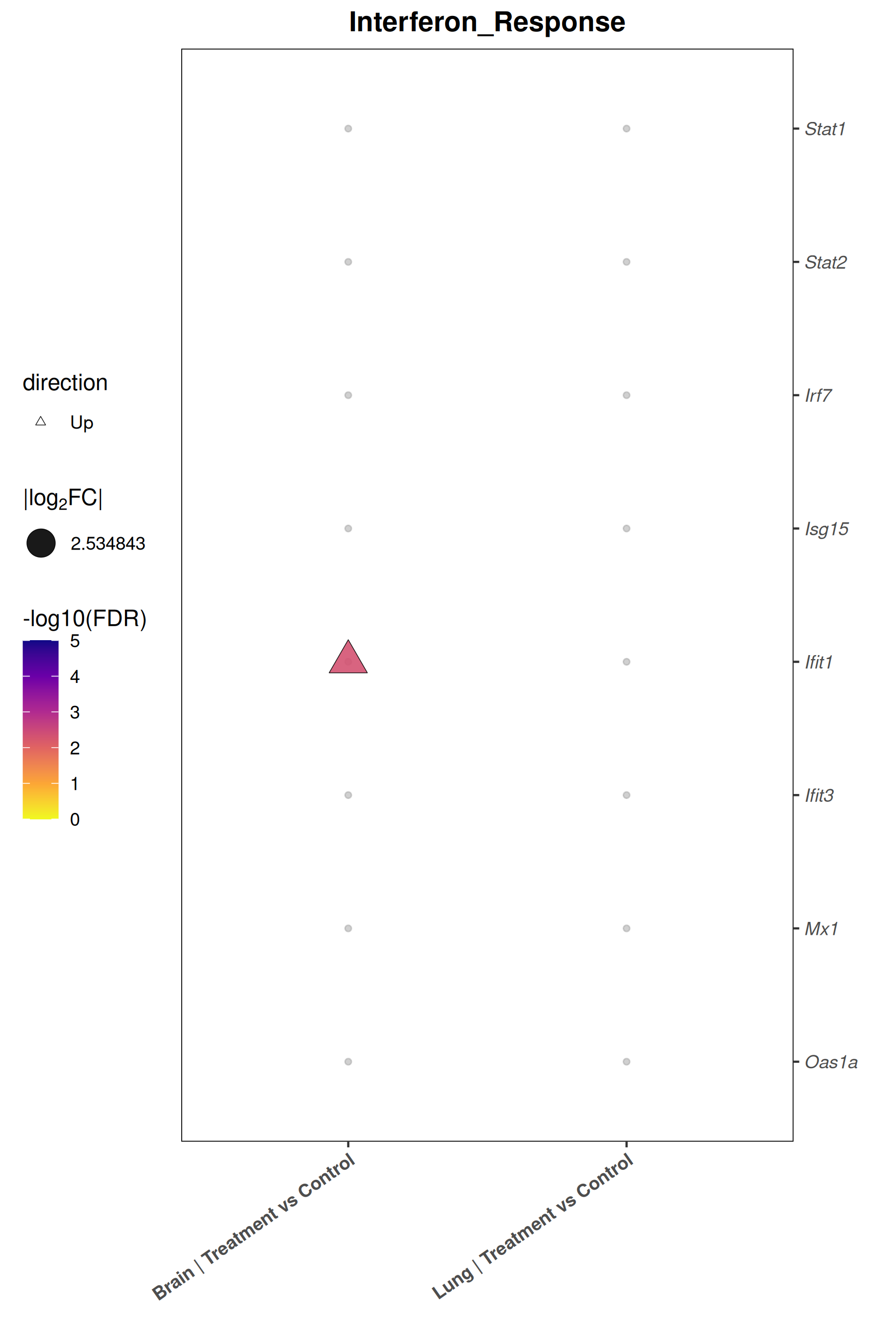
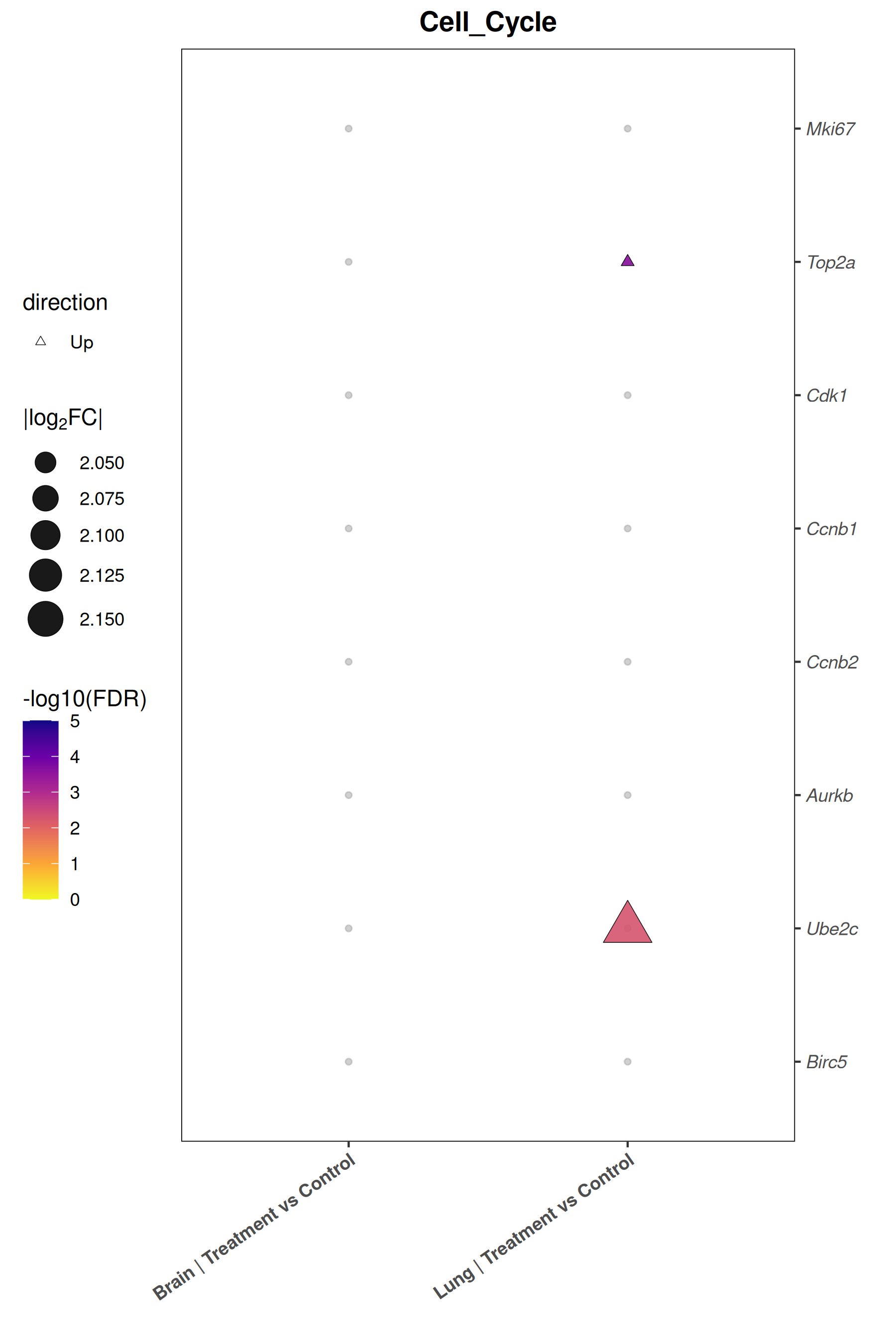
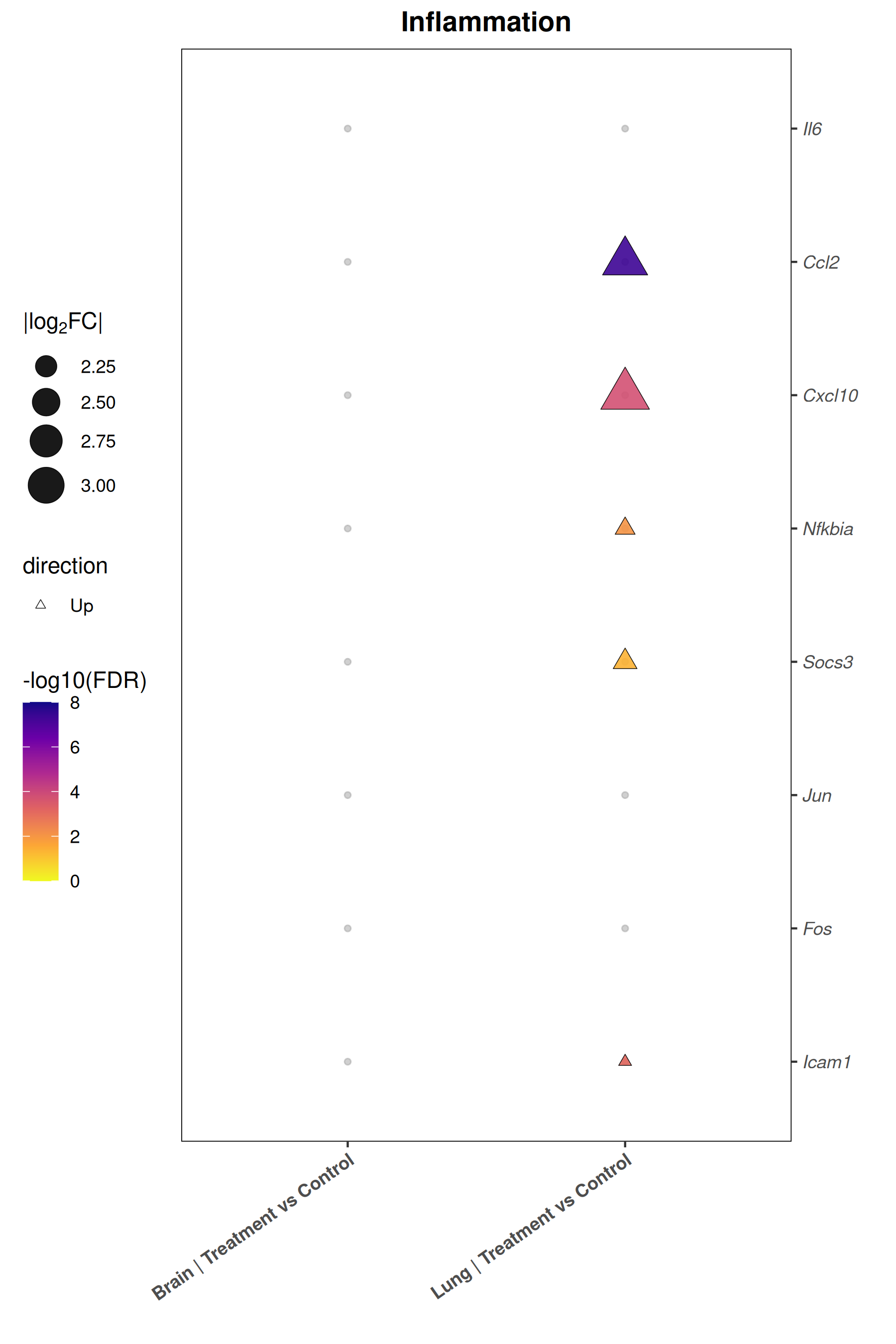
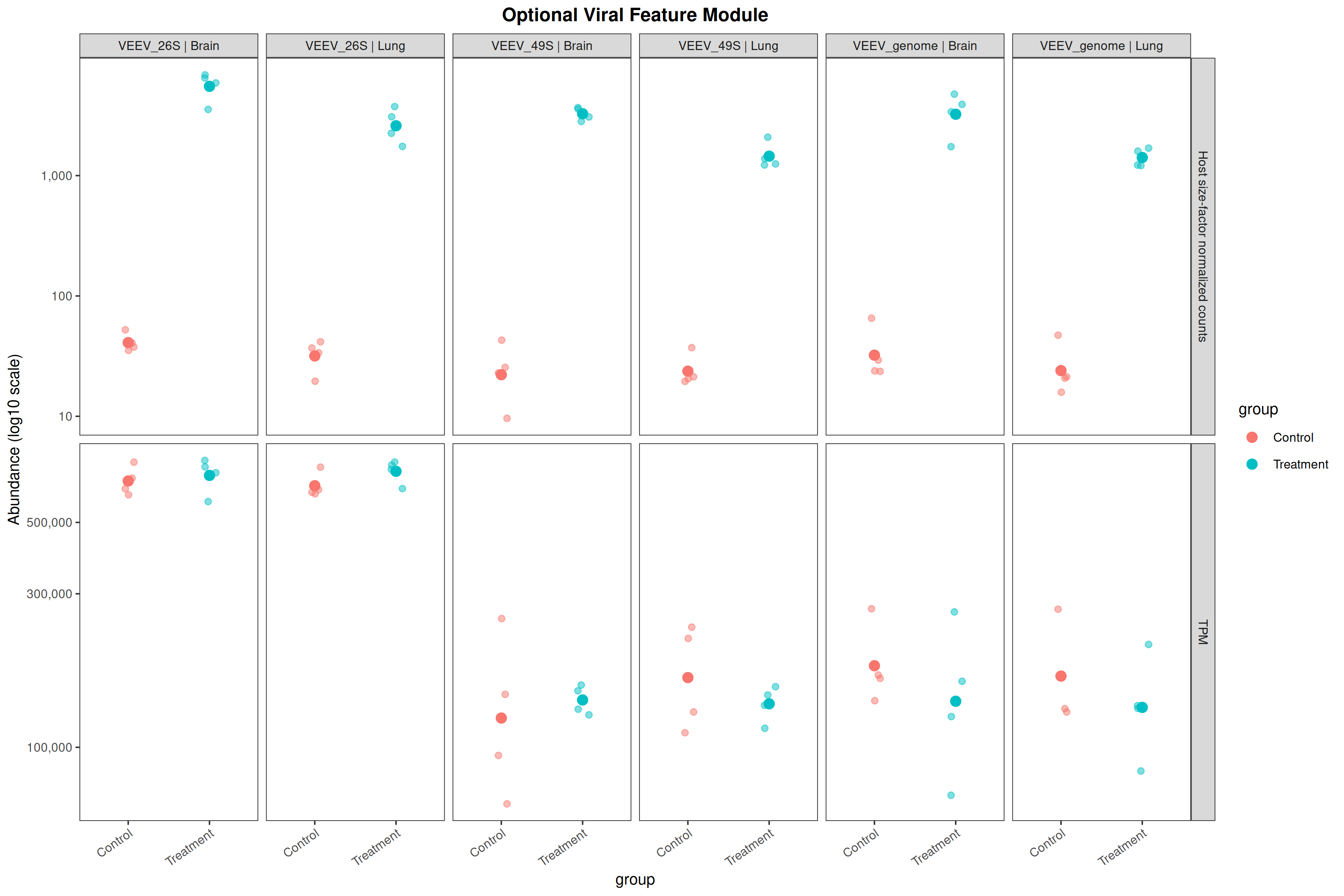
Runs an RNA-seq differential expression workflow inside the notebook. Supports raw count matrices plus metadata or prebuilt SummarizedExperiment inputs, then produces QC diagnostics, DESeq2 contrasts, volcano and MA plots, PCA, top-gene heatmaps, curated gene-set bubble and triangle plots, optional viral-feature panels, and reusable exports.
Overview
Item
Details
Input
Gene counts + metadata or gene/transcript SummarizedExperiment RDS files
When to use this template: You want one notebook that can start from either a count matrix plus sample metadata or a prebuilt SummarizedExperiment, fit DESeq2, and export both publication-ready figures and reusable result tables.
Note
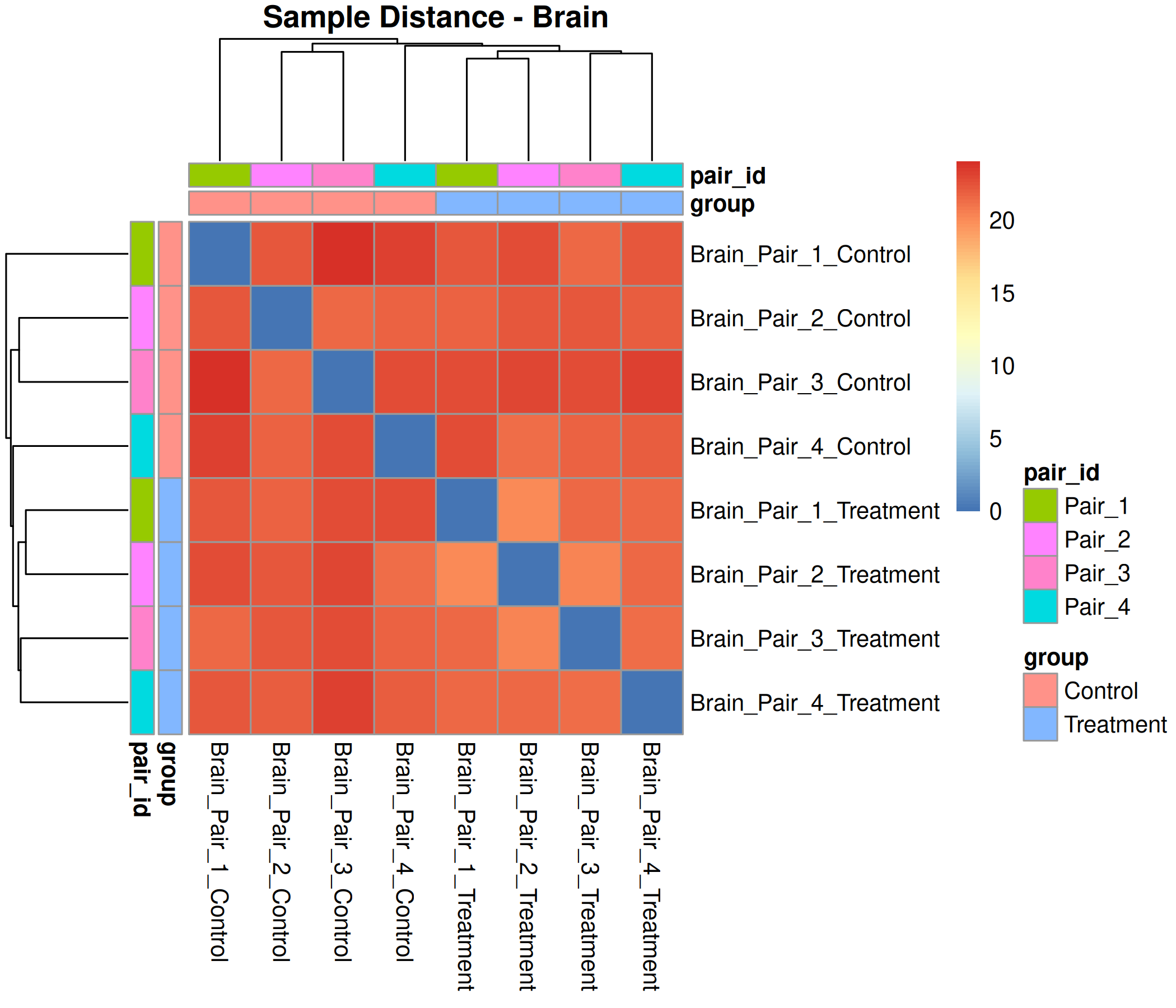
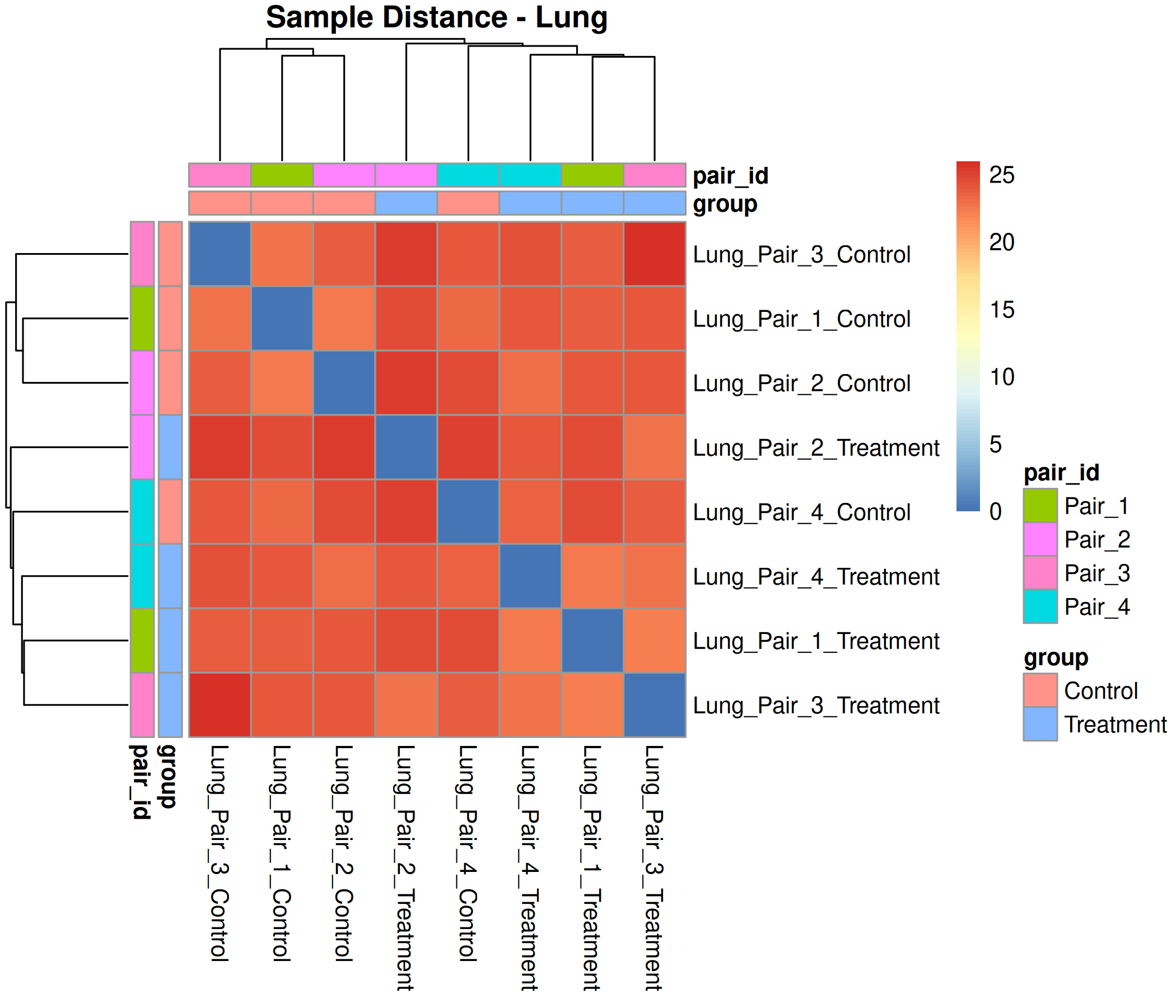
Optional modules: Split-by-subset modeling, blocking terms in the design formula, top-DEG heatmaps, sample-distance heatmaps, and viral transcript abundance panels can all be enabled or disabled from the config block.
---title: "07 · RNA-seq / DESeq2"subtitle: "End-to-end differential expression from counts or SummarizedExperiment inputs"description: | Runs an RNA-seq differential expression workflow inside the notebook. Supports raw count matrices plus metadata or prebuilt SummarizedExperiment inputs, then produces QC diagnostics, DESeq2 contrasts, volcano and MA plots, PCA, top-gene heatmaps, curated gene-set bubble and triangle plots, optional viral-feature panels, and reusable exports.categories: [transcriptomics, RNA-seq, DESeq2, differential-expression]---## Overview| Item | Details ||------|---------|| **Input** | Gene counts + metadata or gene/transcript `SummarizedExperiment` RDS files || **Key packages** | `DESeq2`, `SummarizedExperiment`, `ggplot2`, `patchwork`, `pheatmap` || **Statistics** | DESeq2 Wald test · FDR-adjusted p-values (BH) || **Output** | Diagnostics · PCA · Volcano/MA plots · Heatmaps · Bubble/Triangle plots · DEG tables · Analysis bundle || **Download** | [template.Rmd](template.Rmd) |::: {.callout-tip}**When to use this template:** You want one notebook that can start from either acount matrix plus sample metadata or a prebuilt SummarizedExperiment, fit DESeq2,and export both publication-ready figures and reusable result tables.:::::: {.callout-note}**Optional modules:** Split-by-subset modeling, blocking terms in the design formula,top-DEG heatmaps, sample-distance heatmaps, and viral transcript abundance panels canall be enabled or disabled from the config block.:::[Back to Gallery](../../index.html){.btn .btn-outline-secondary}[Open Template File](template.Rmd){.btn .btn-primary}```{r child="template.Rmd"}```