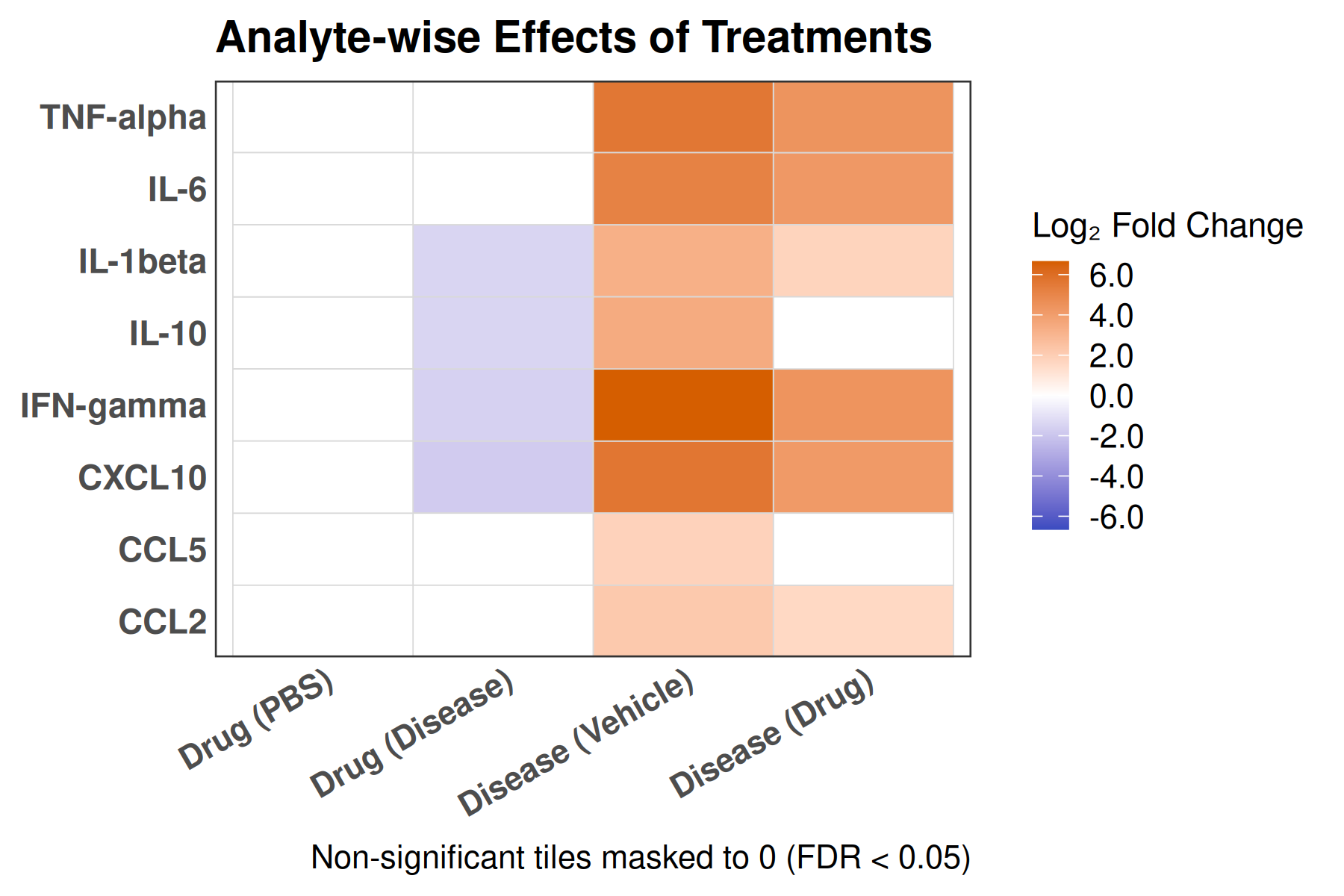
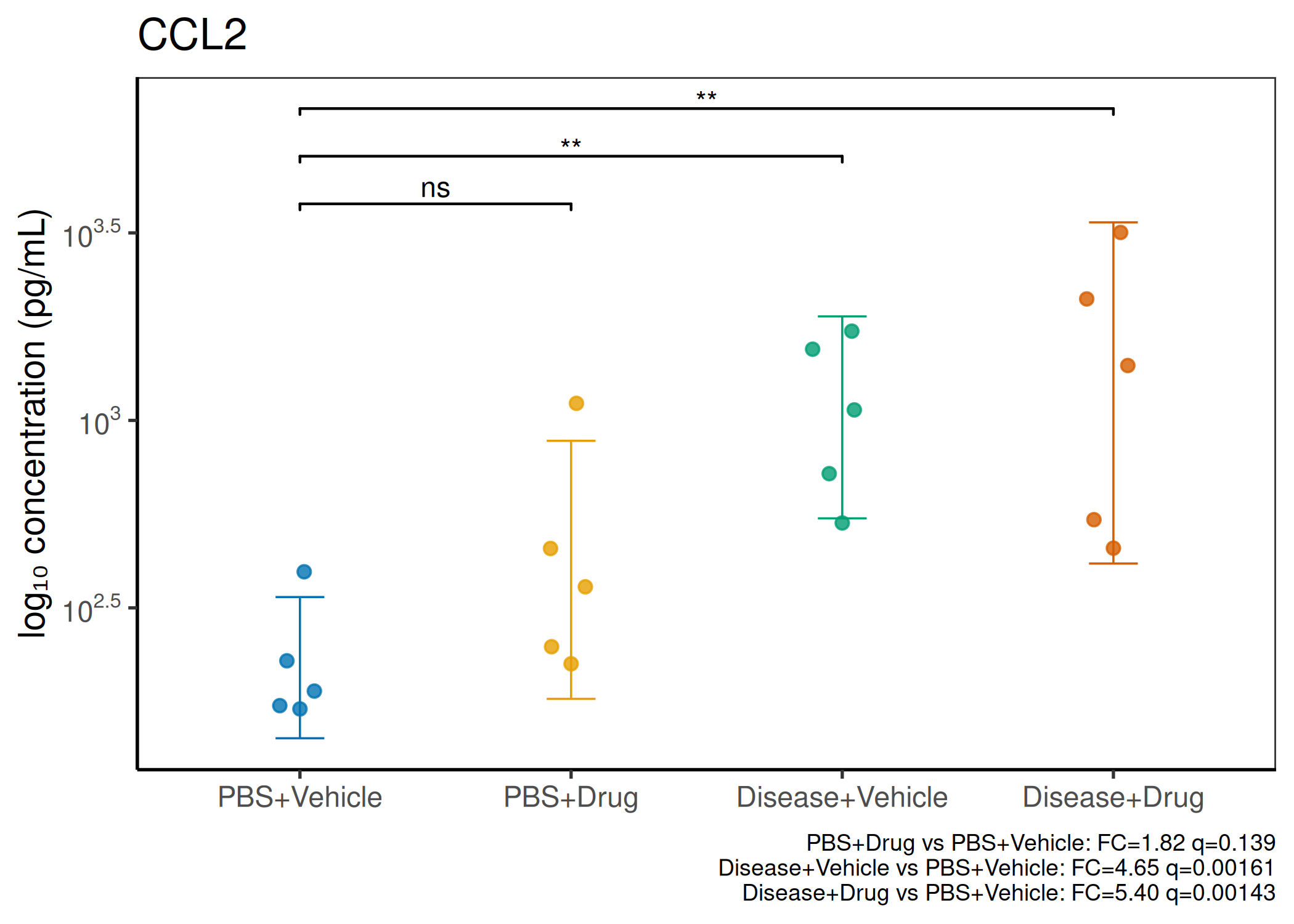
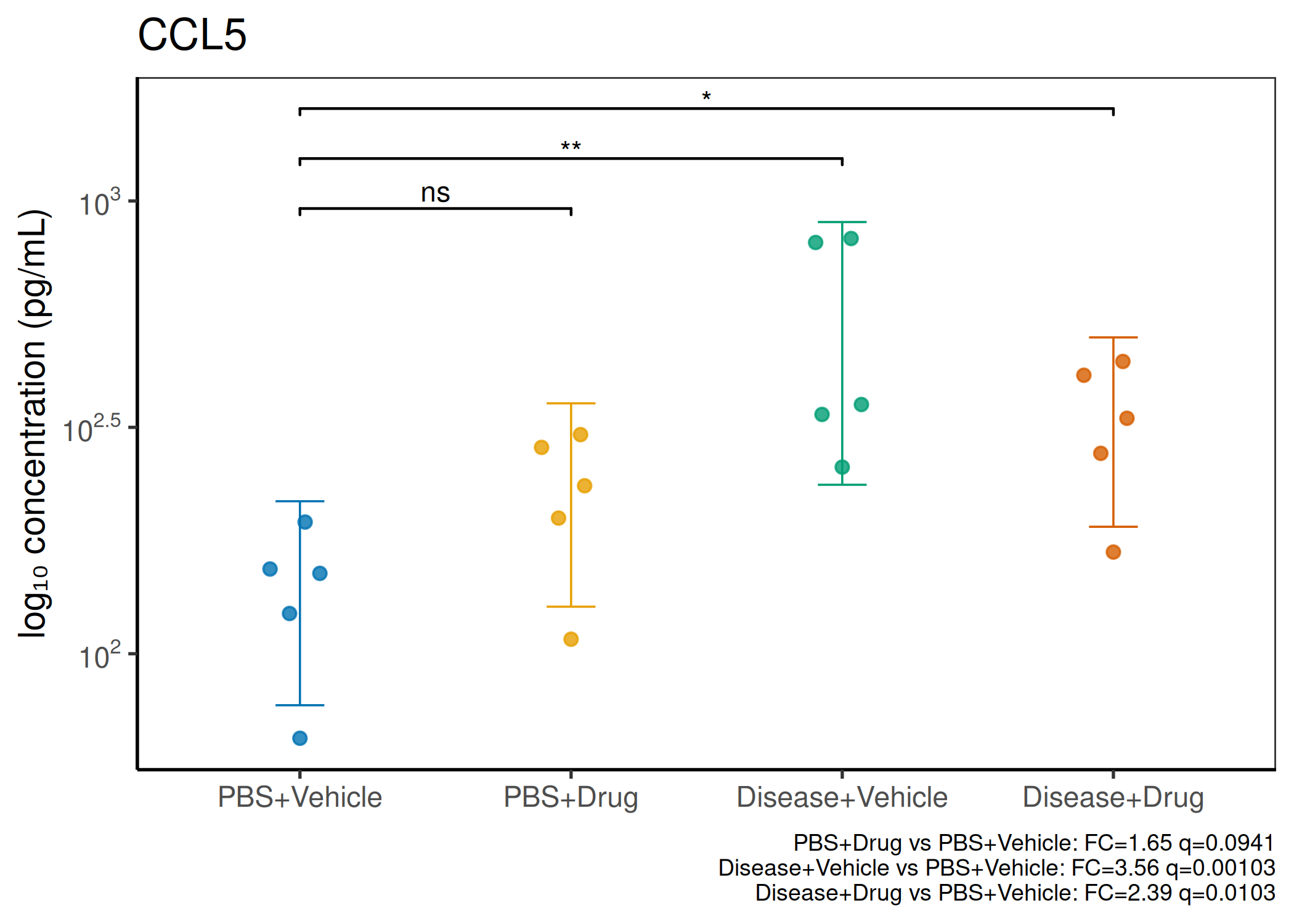
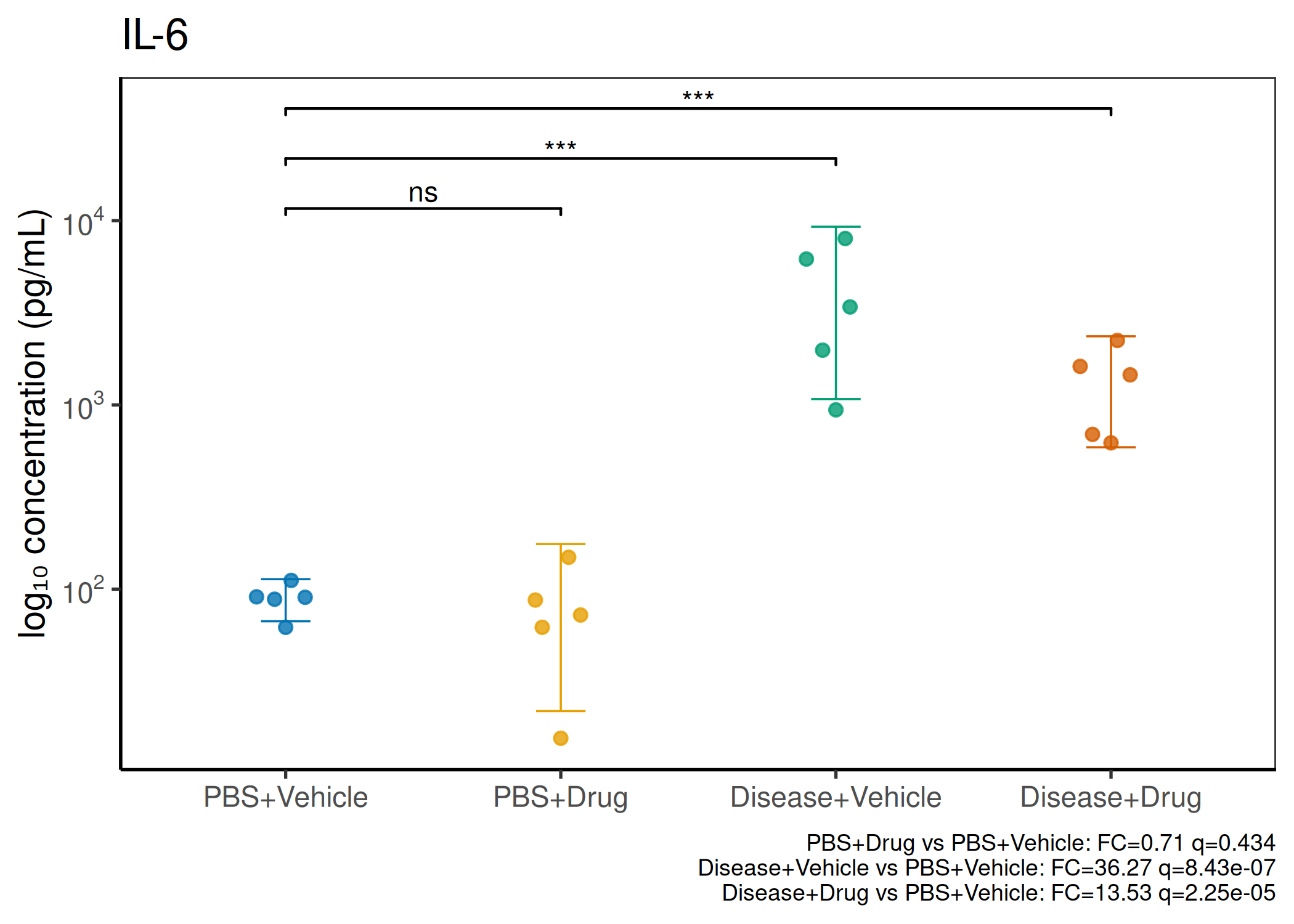
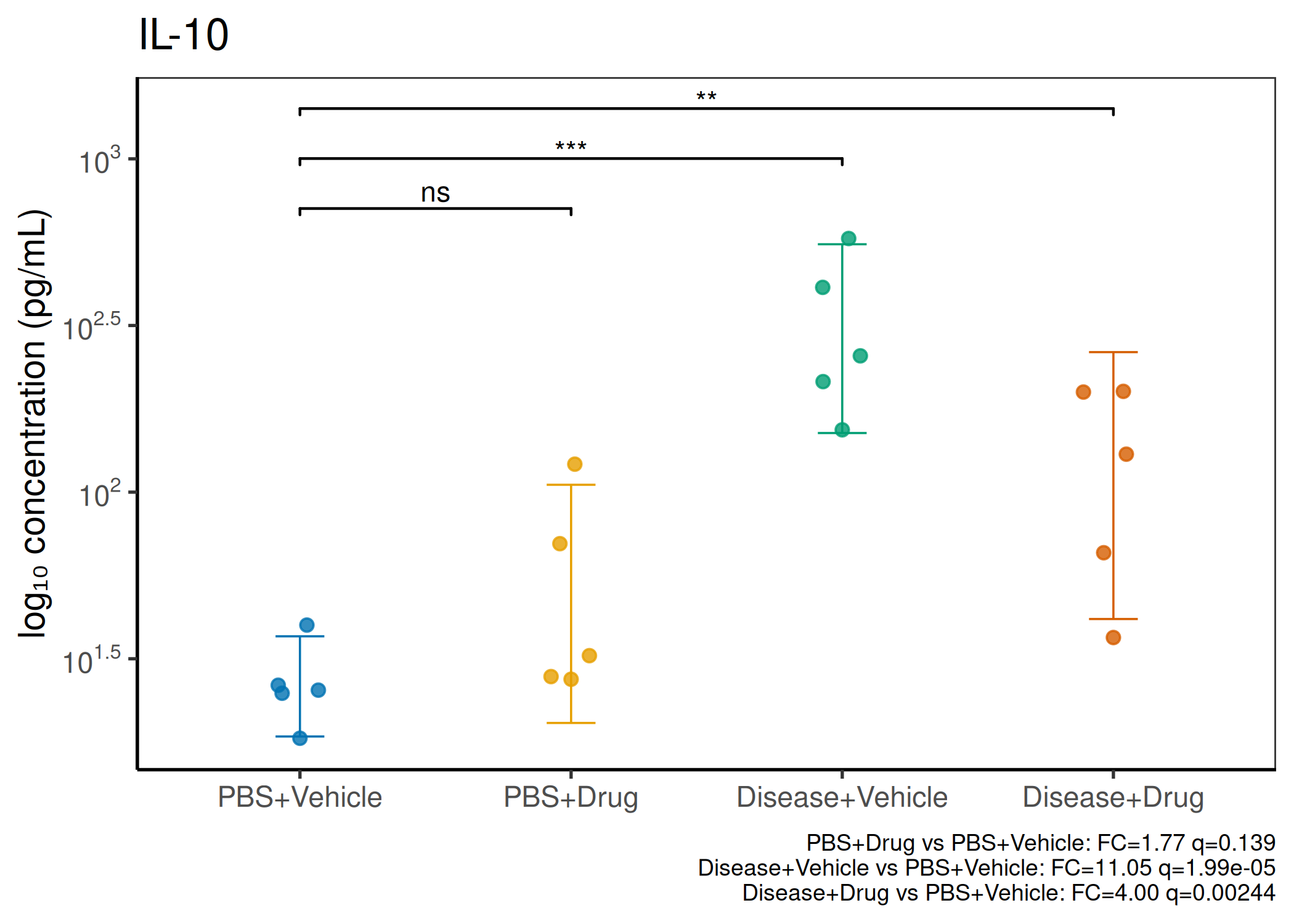
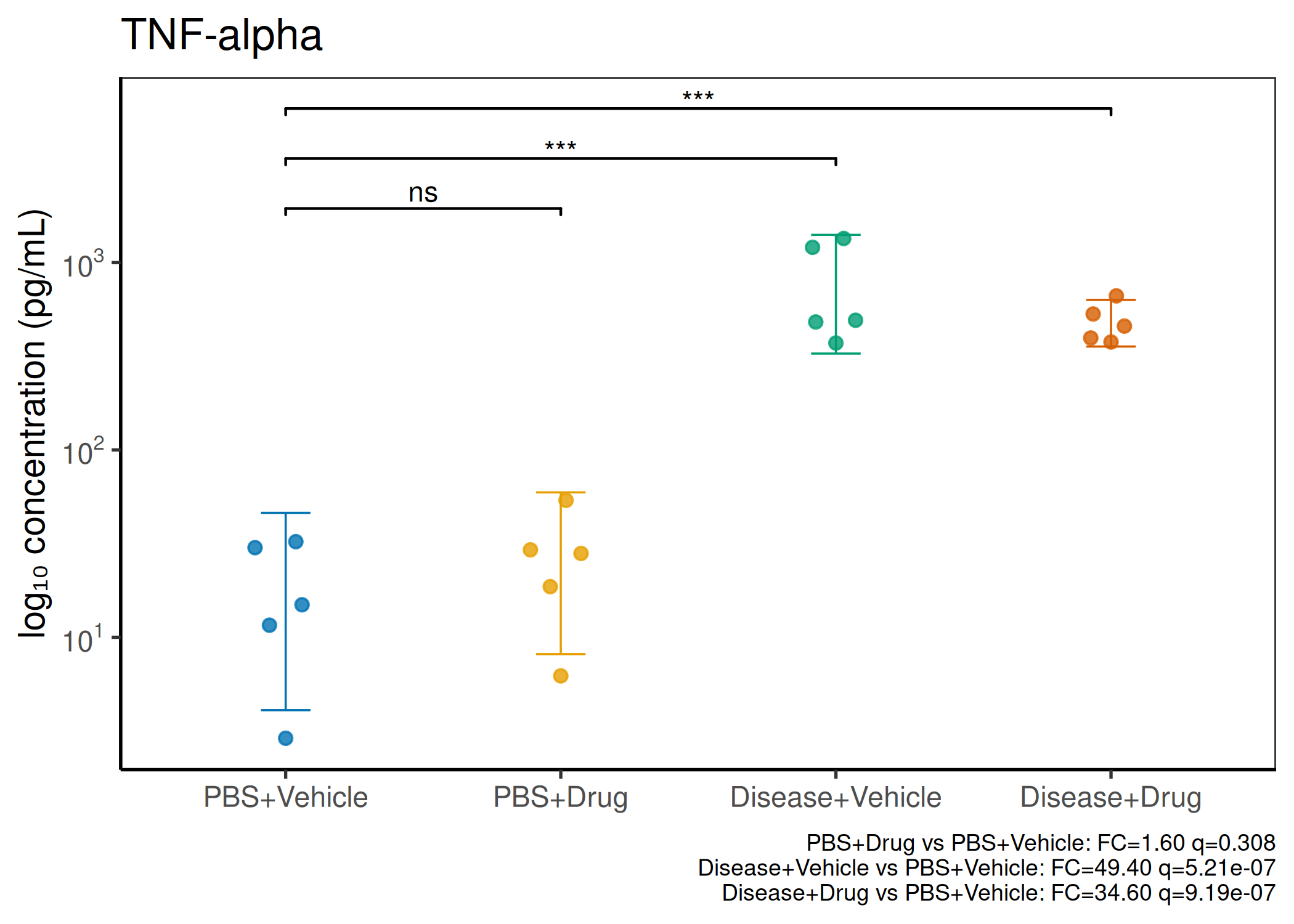
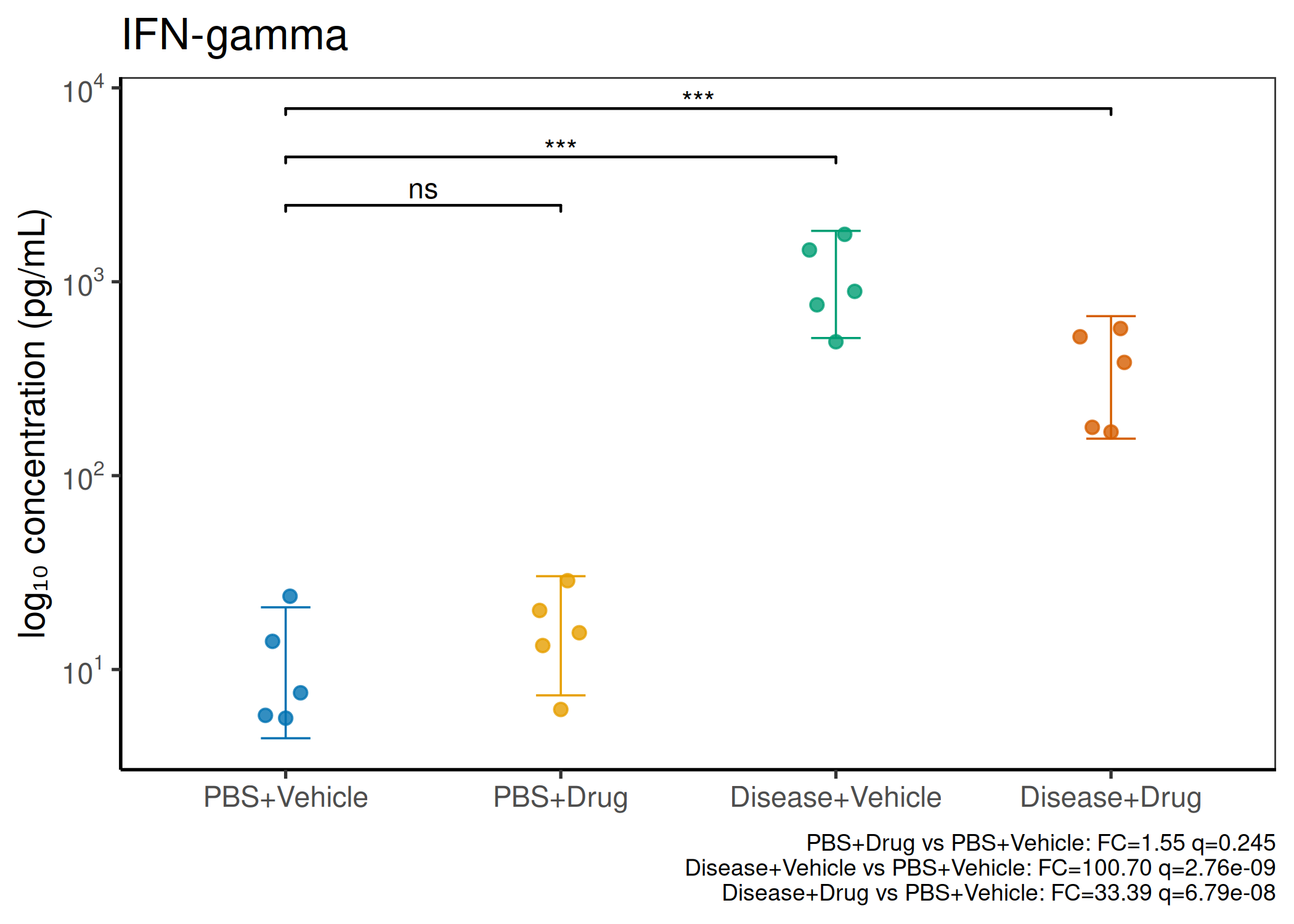
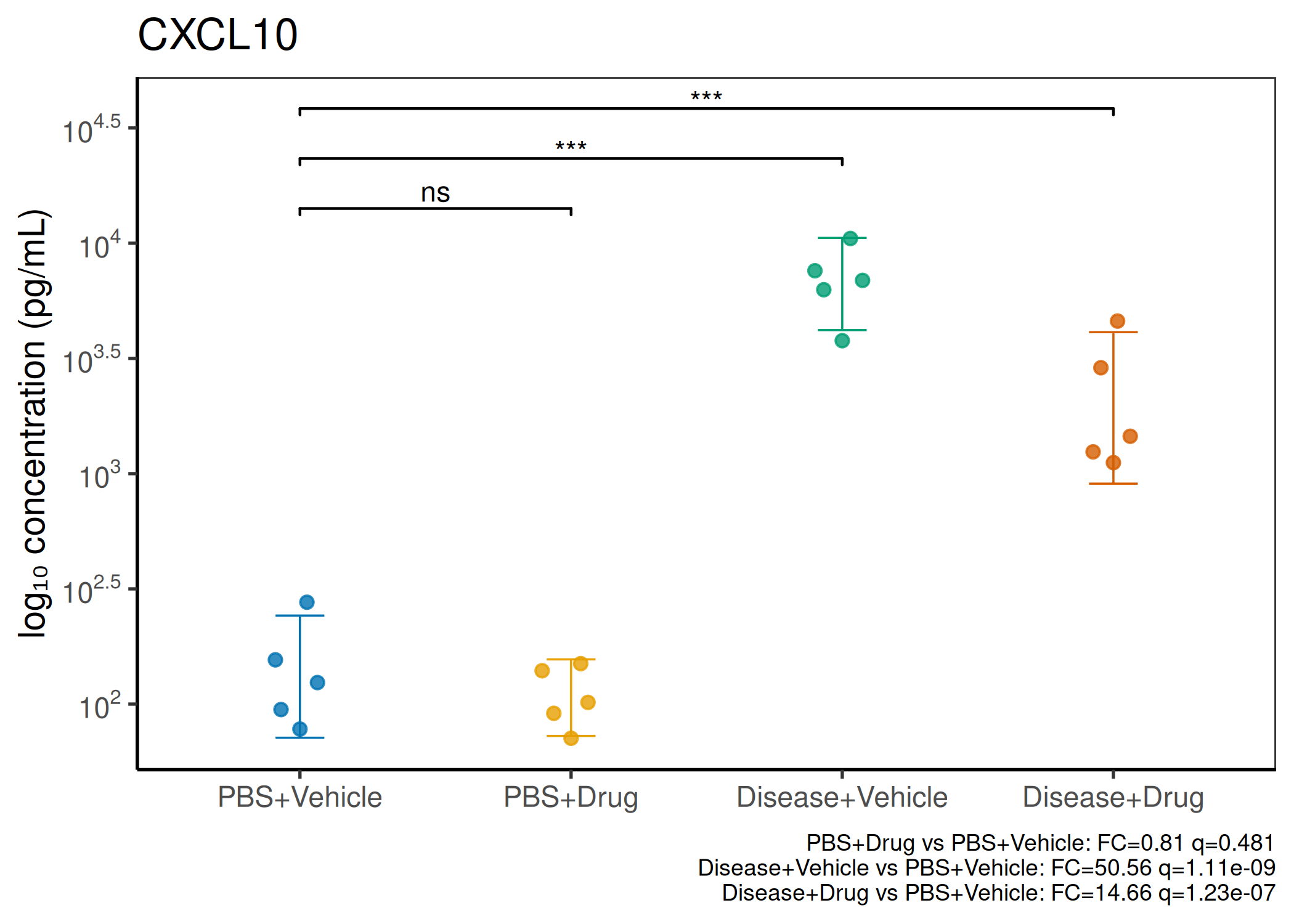
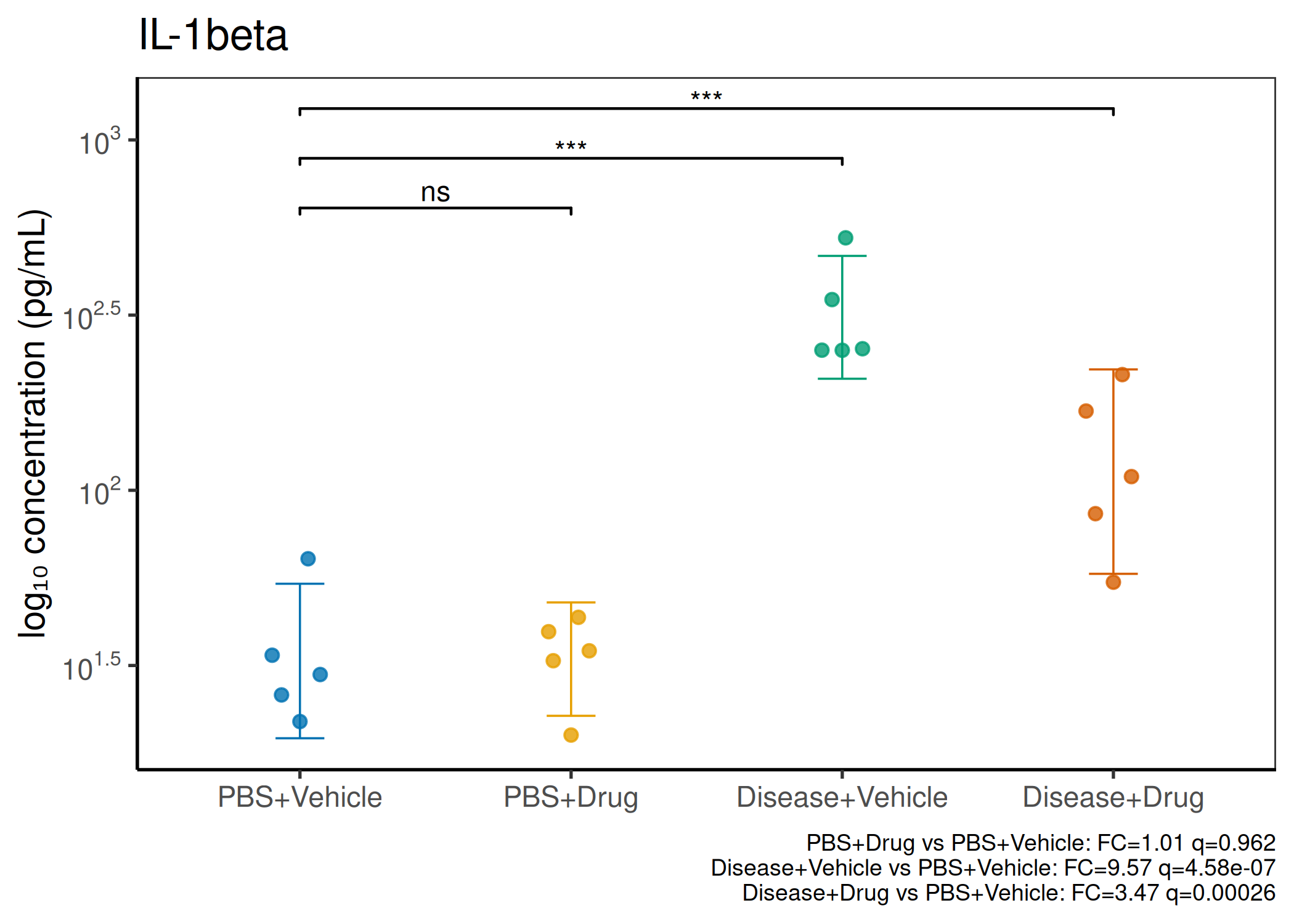
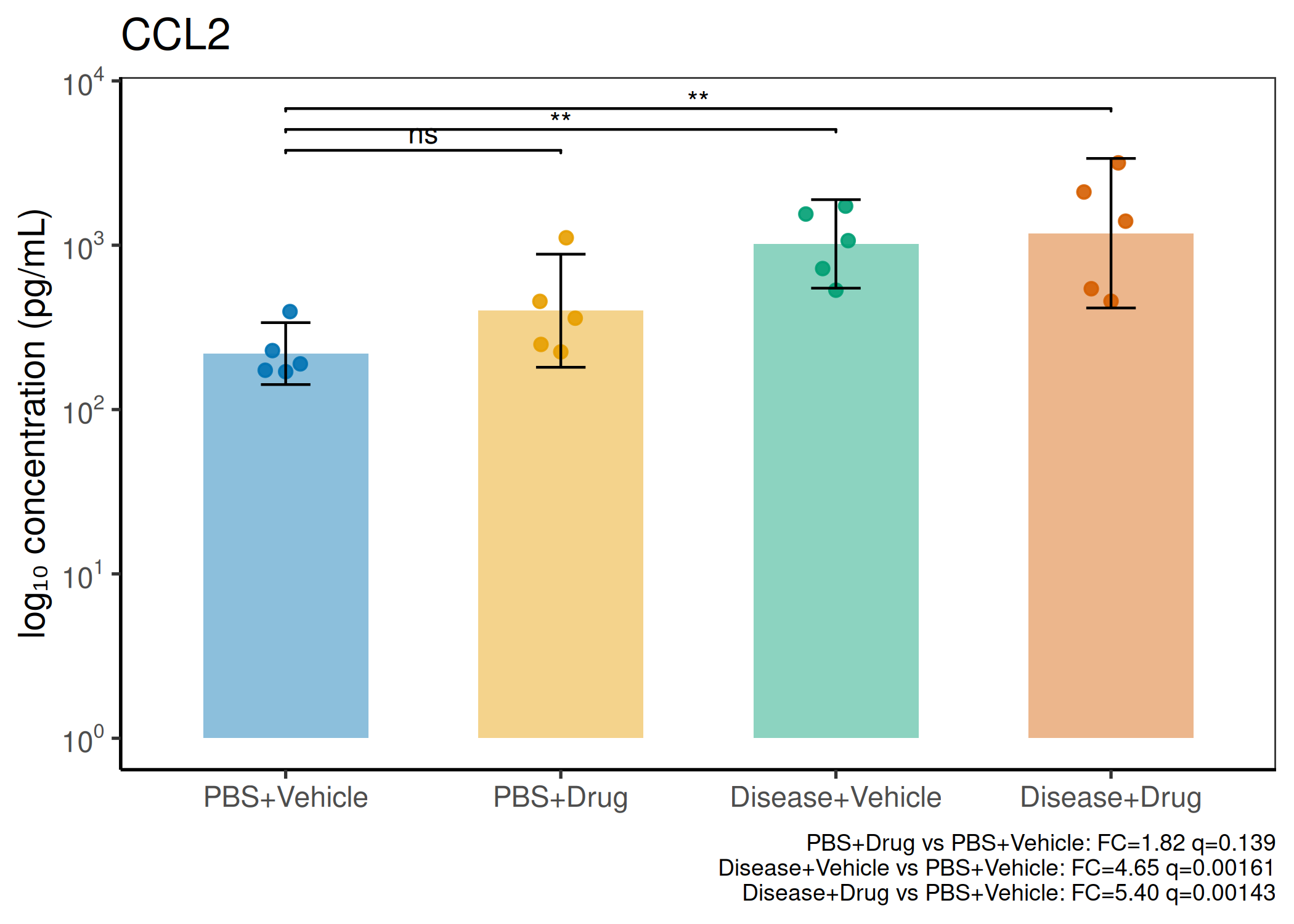
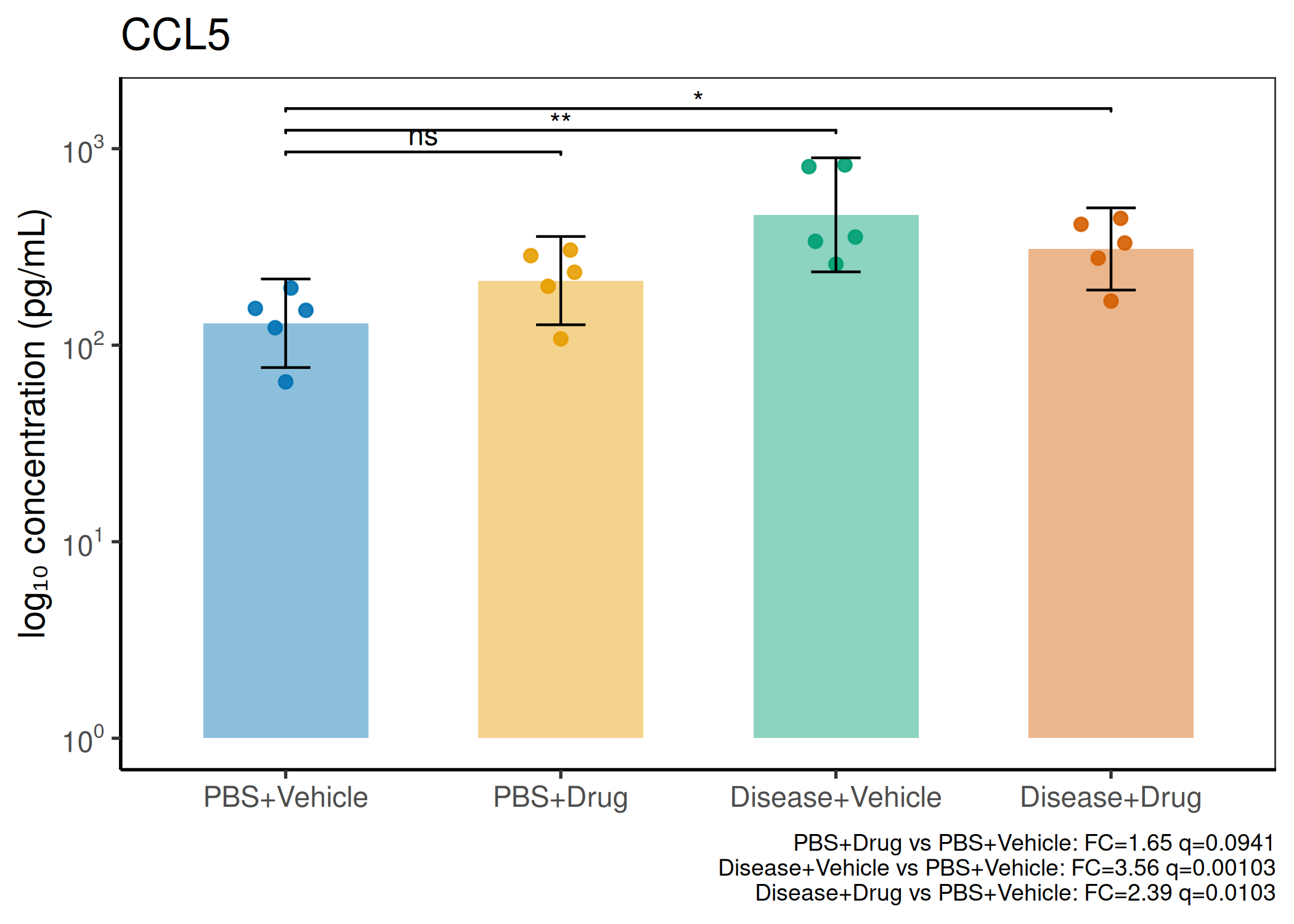
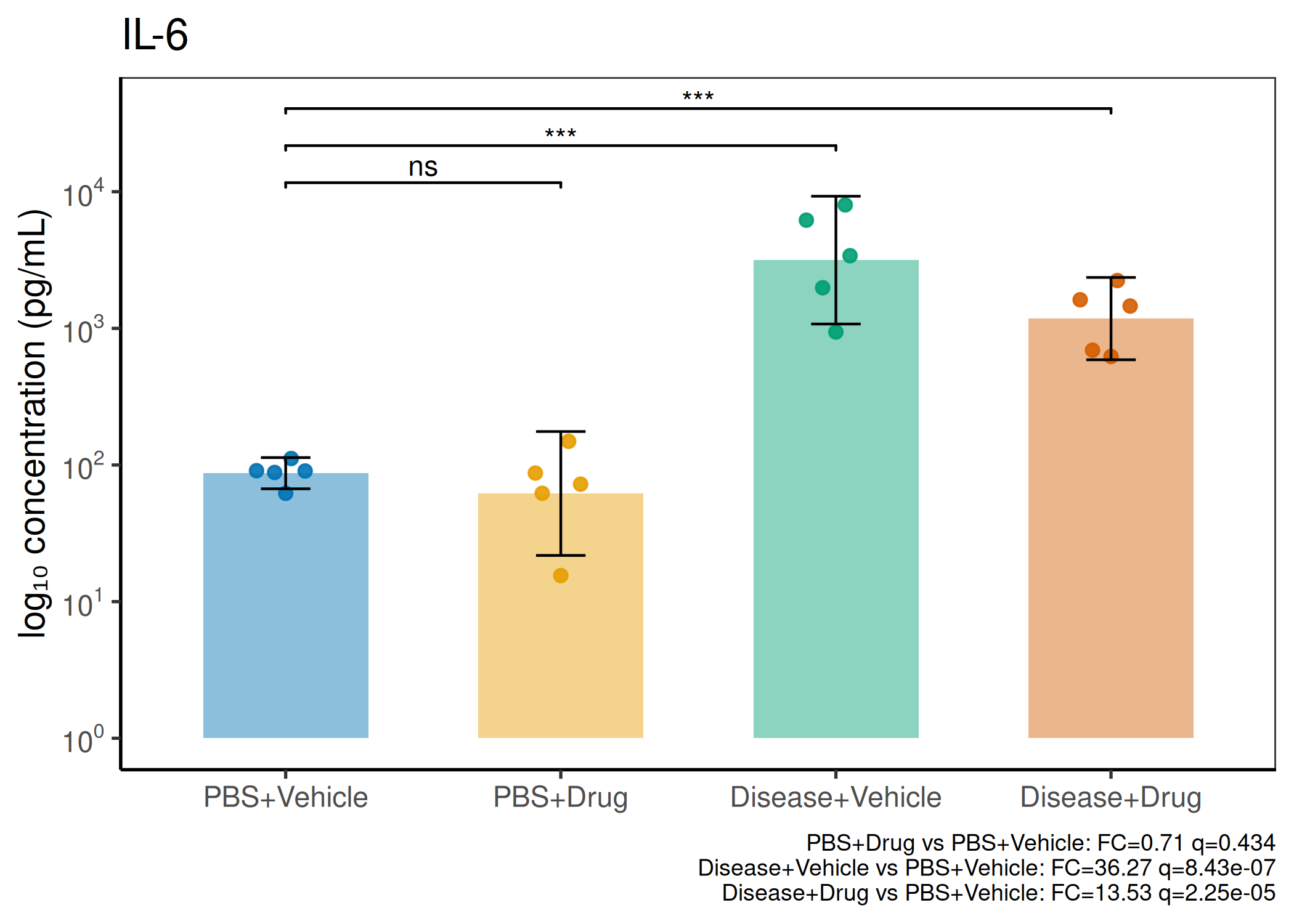
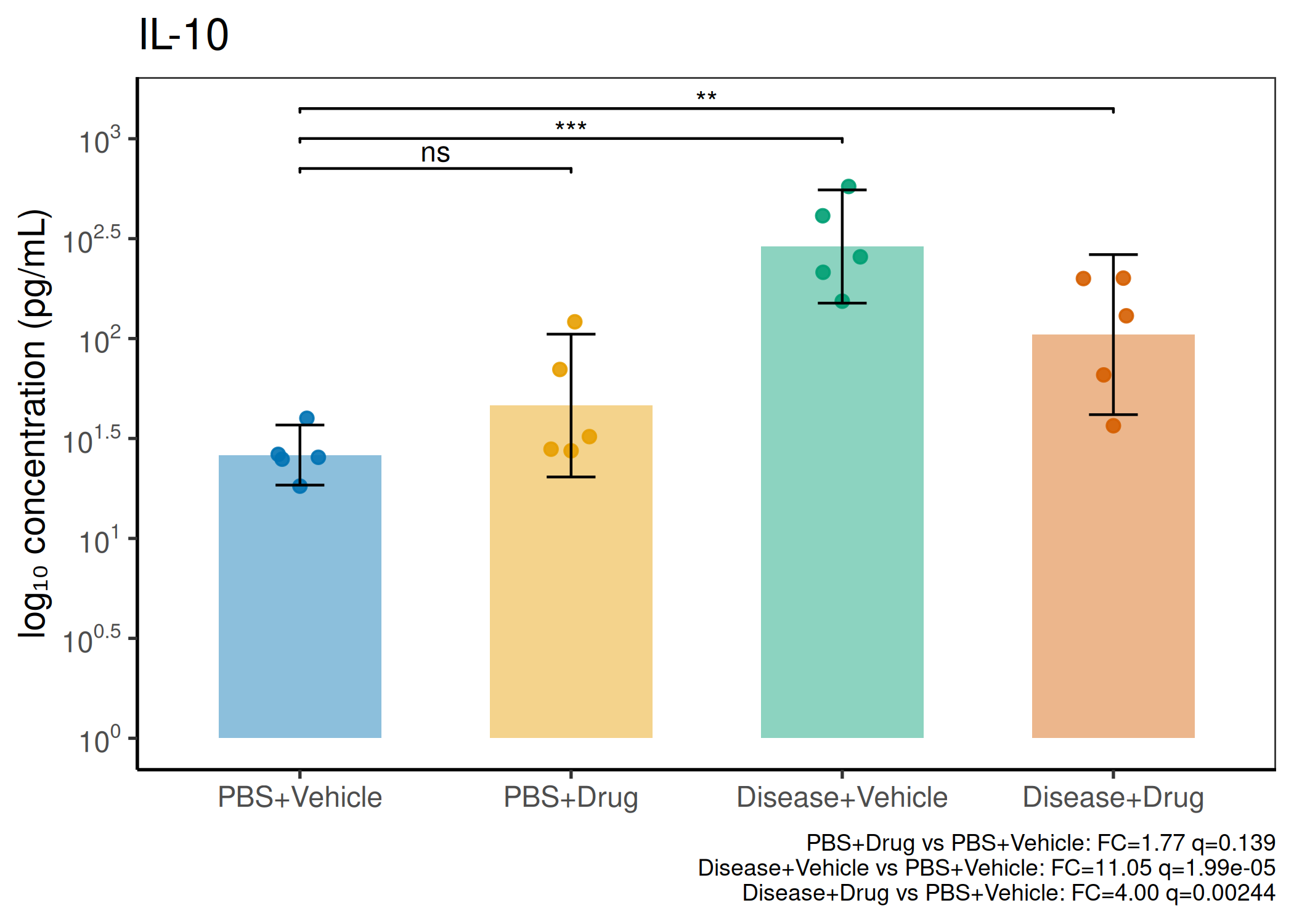
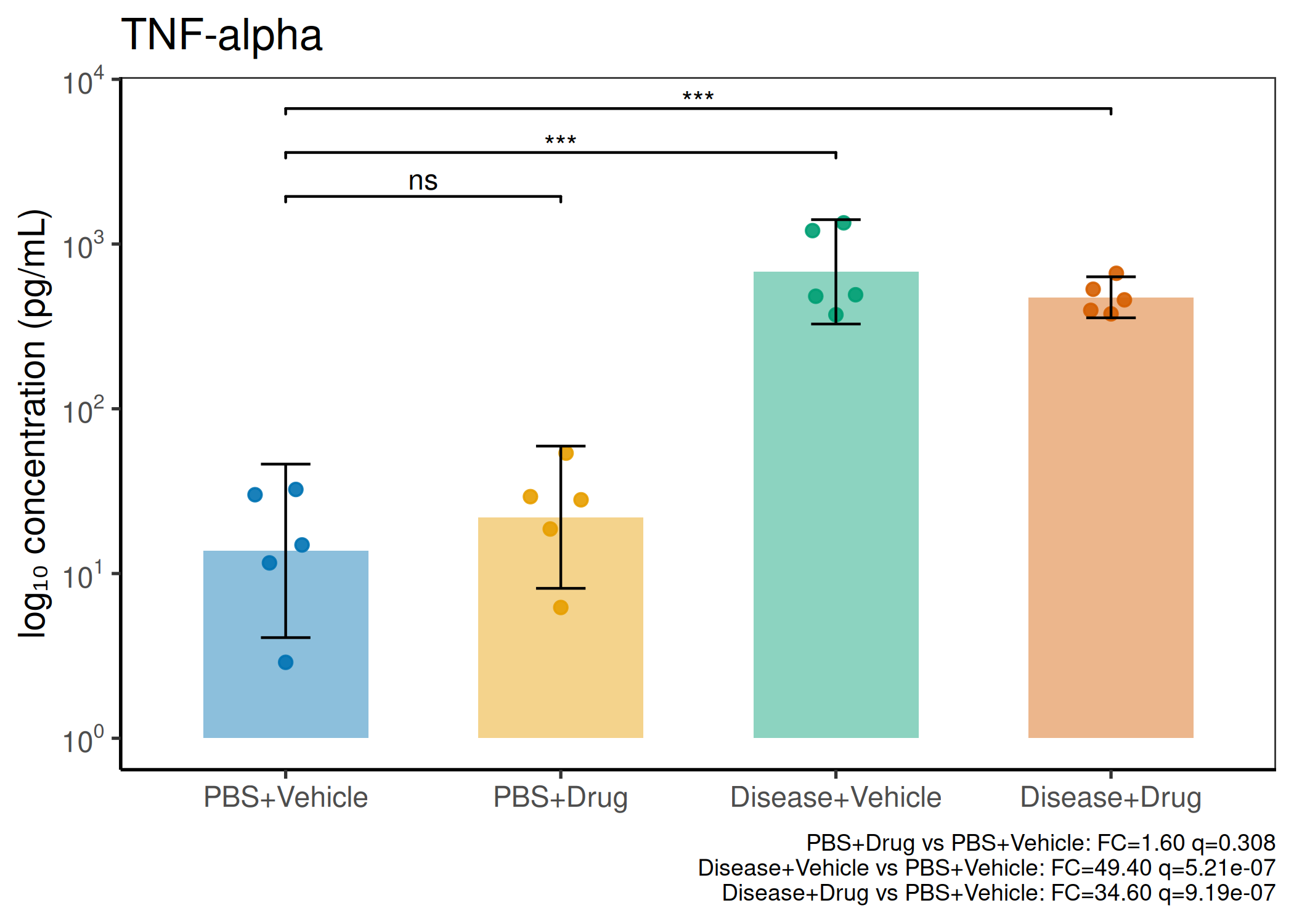
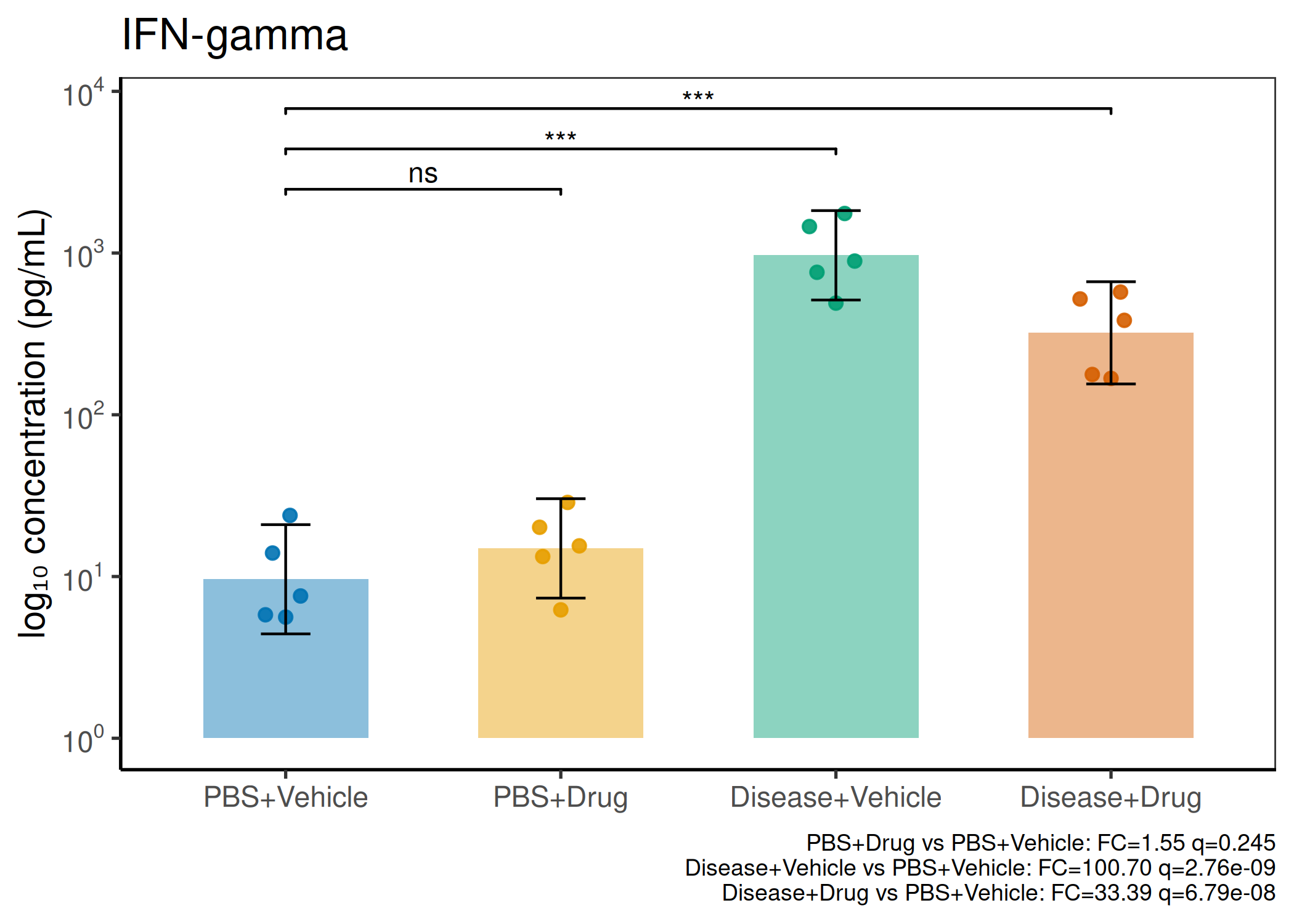
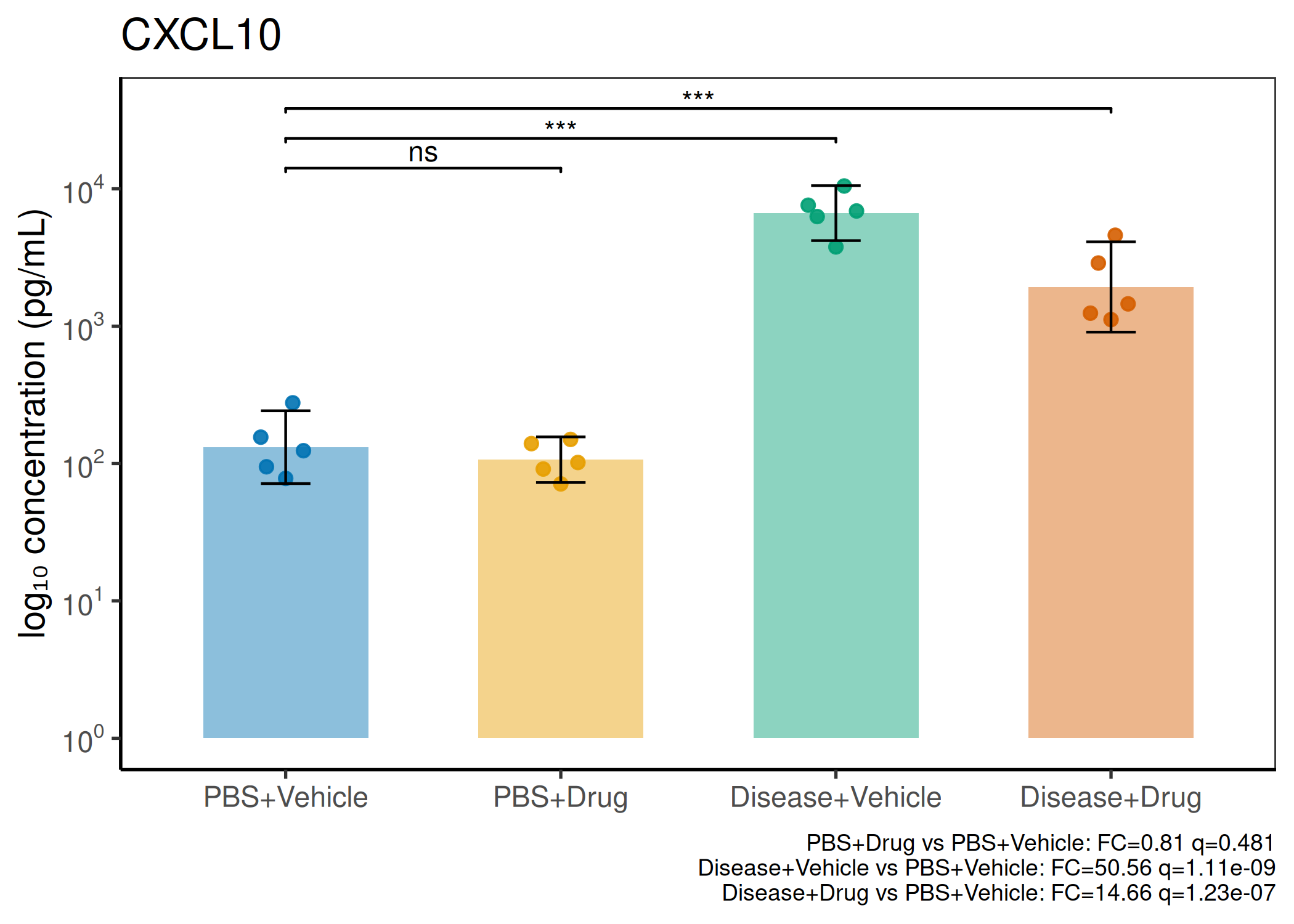
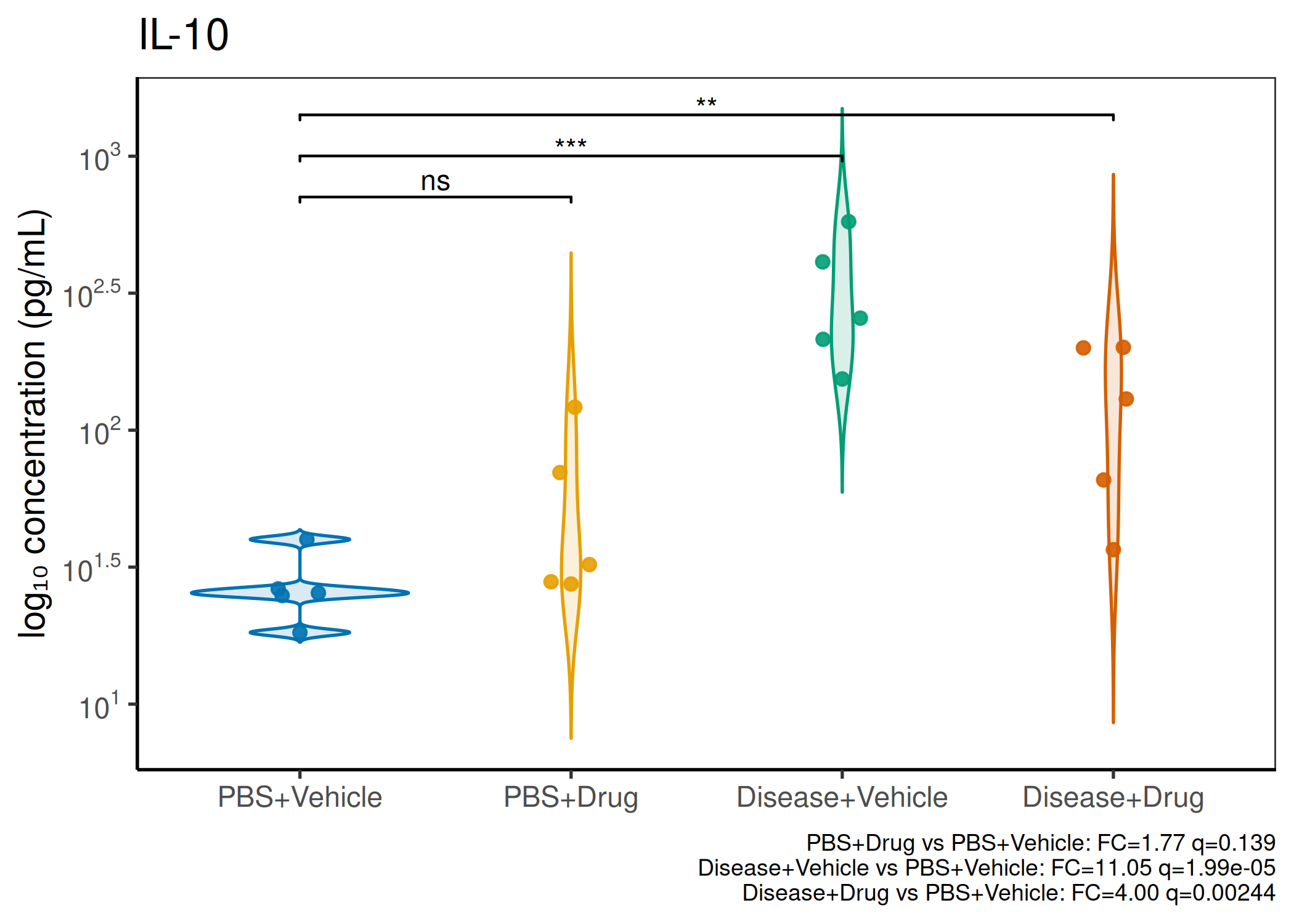
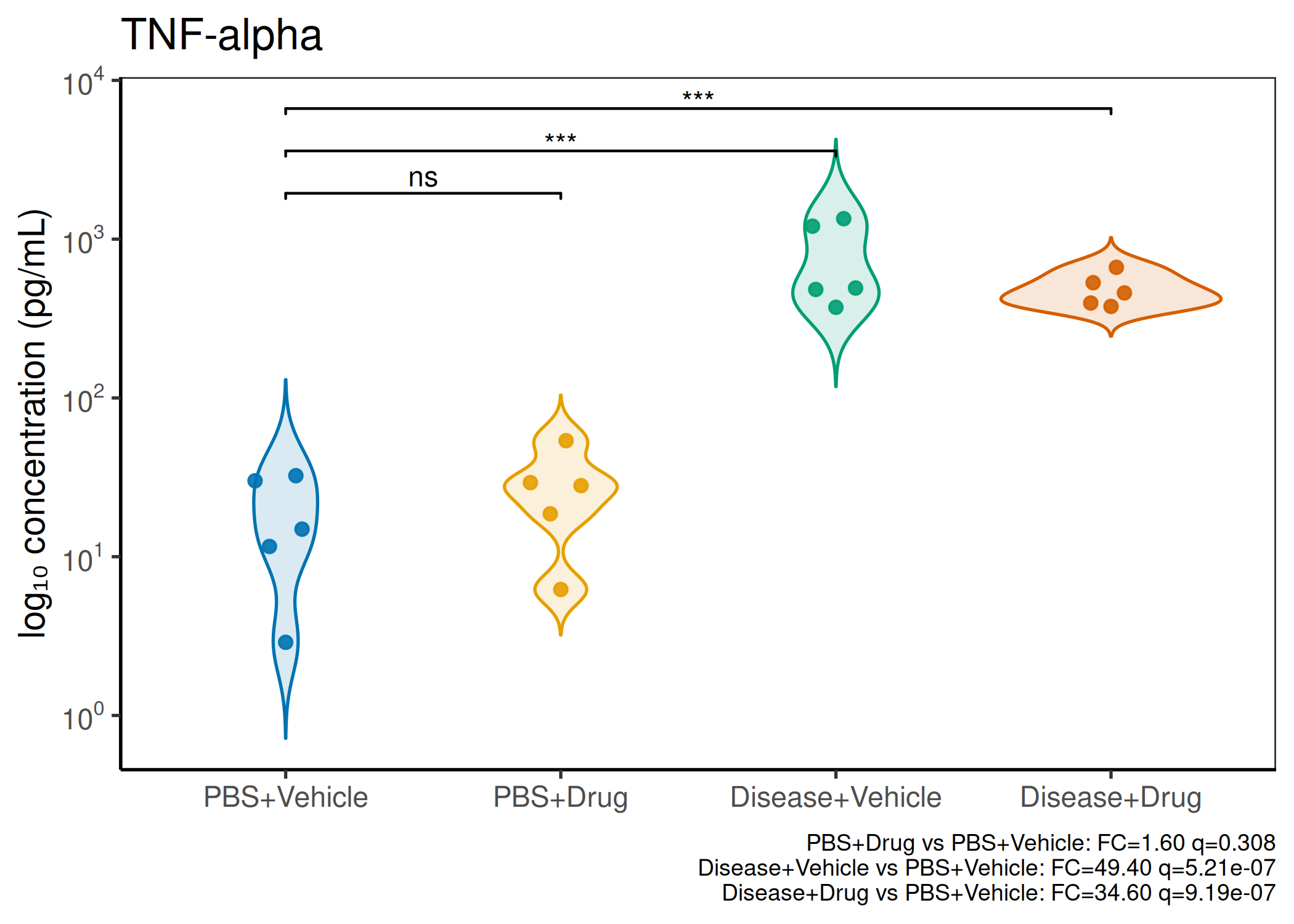
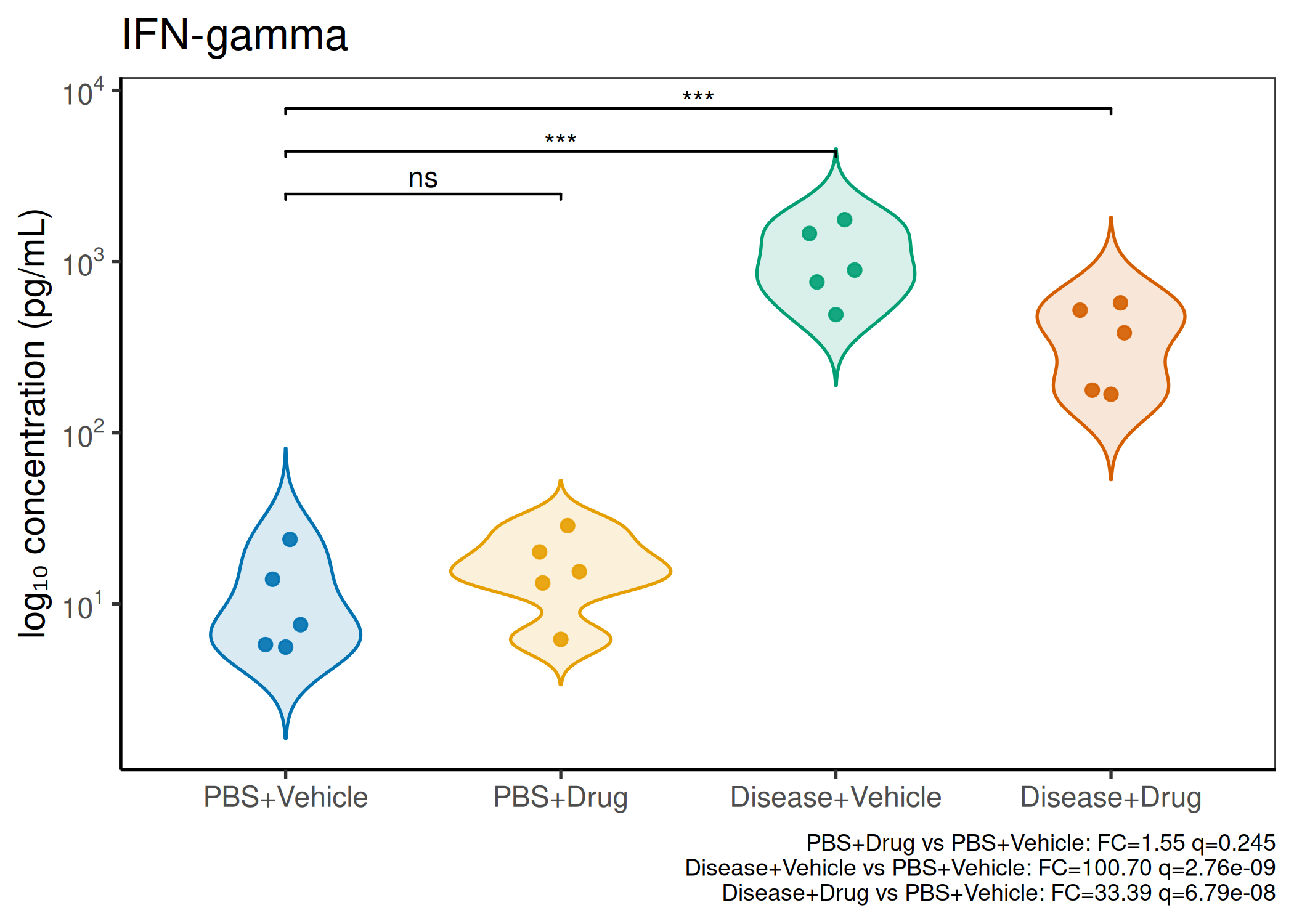
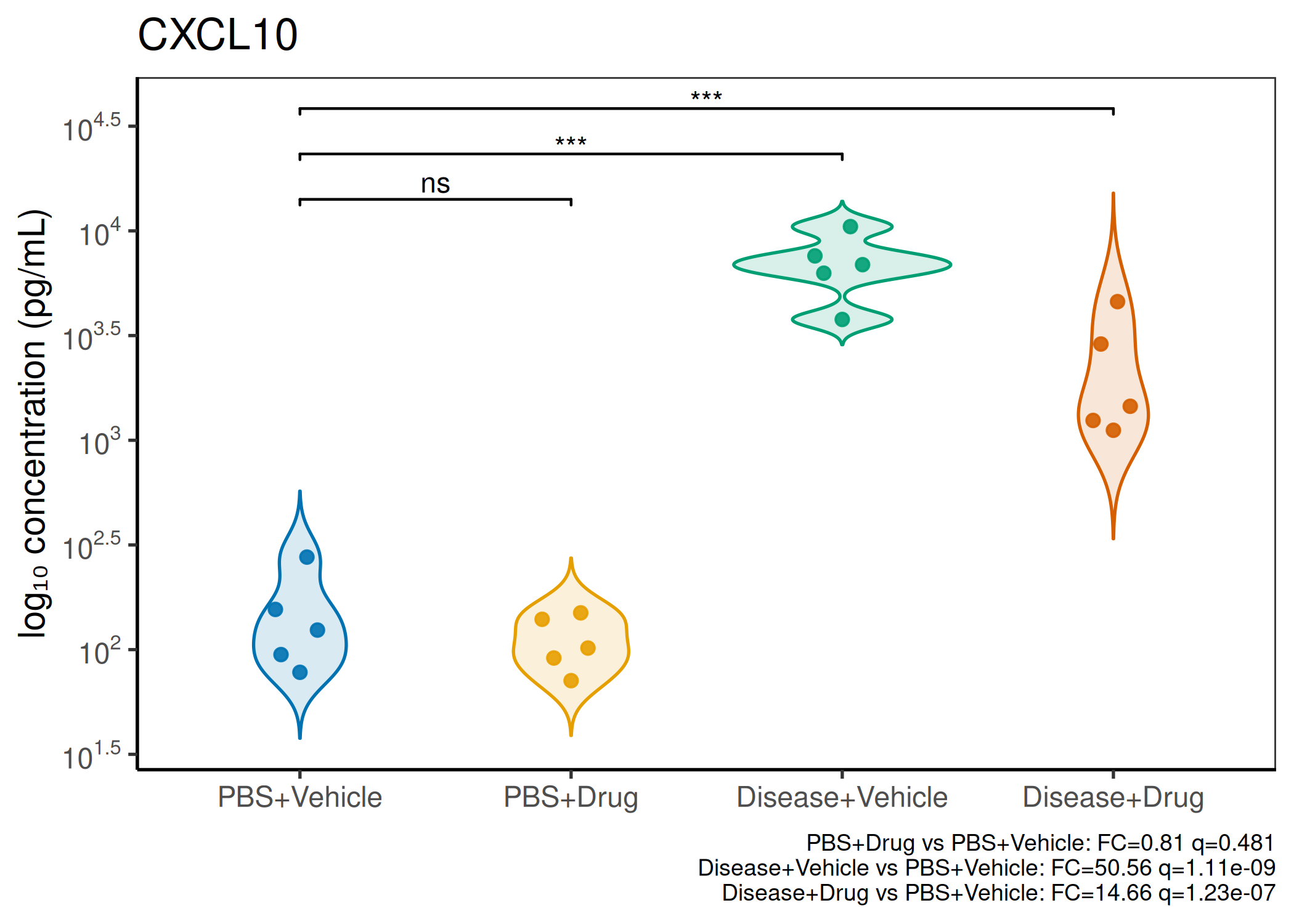
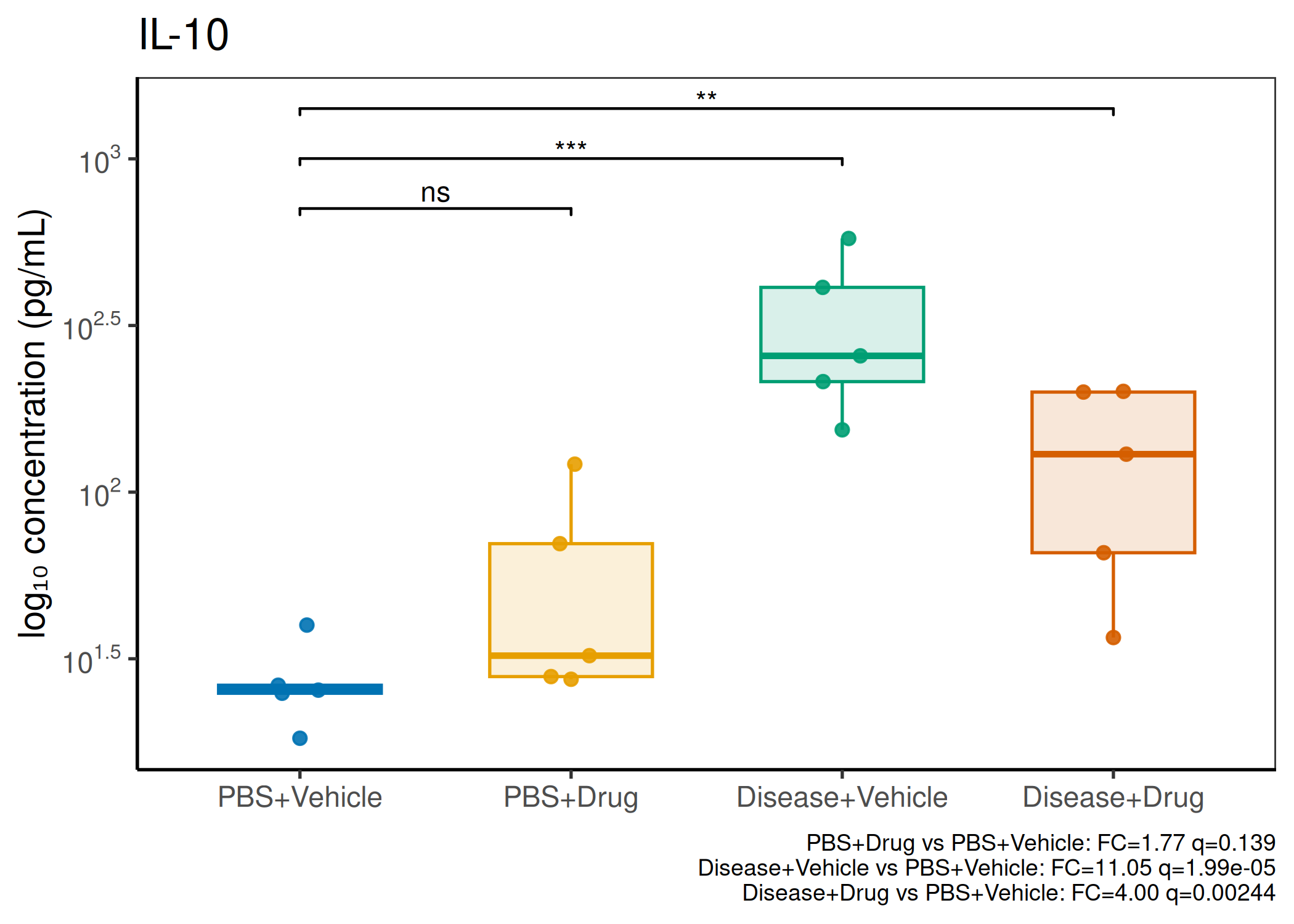
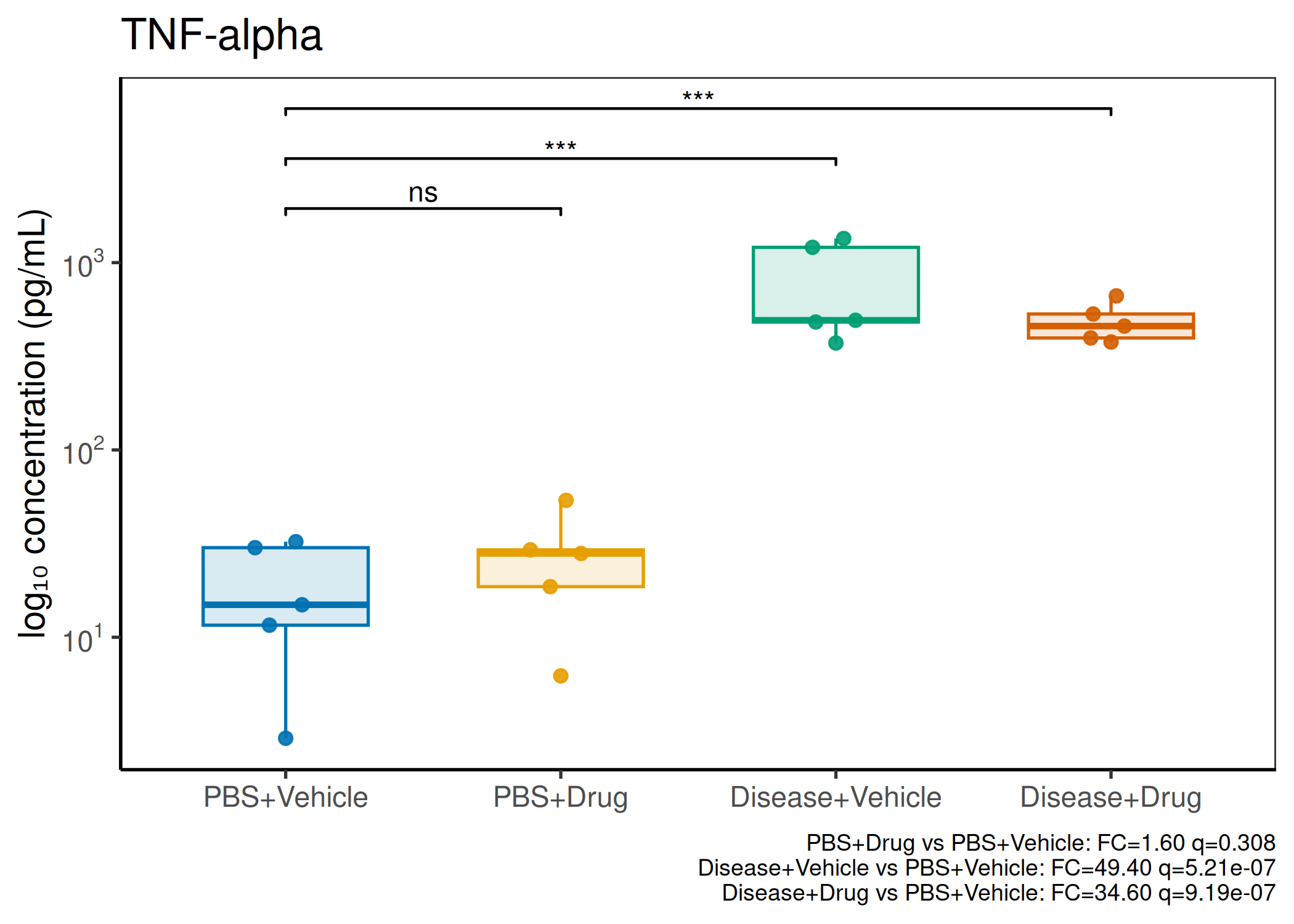
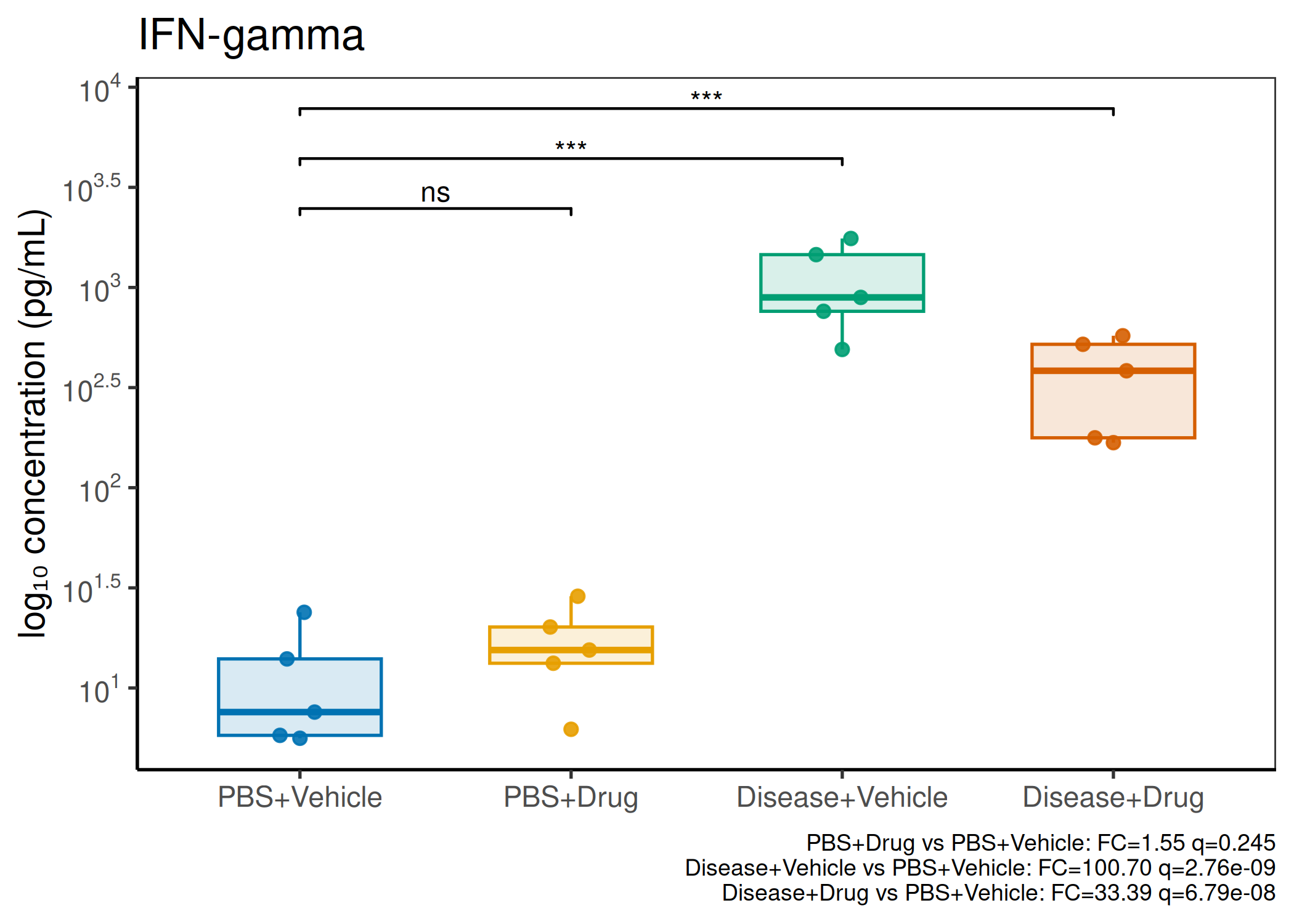
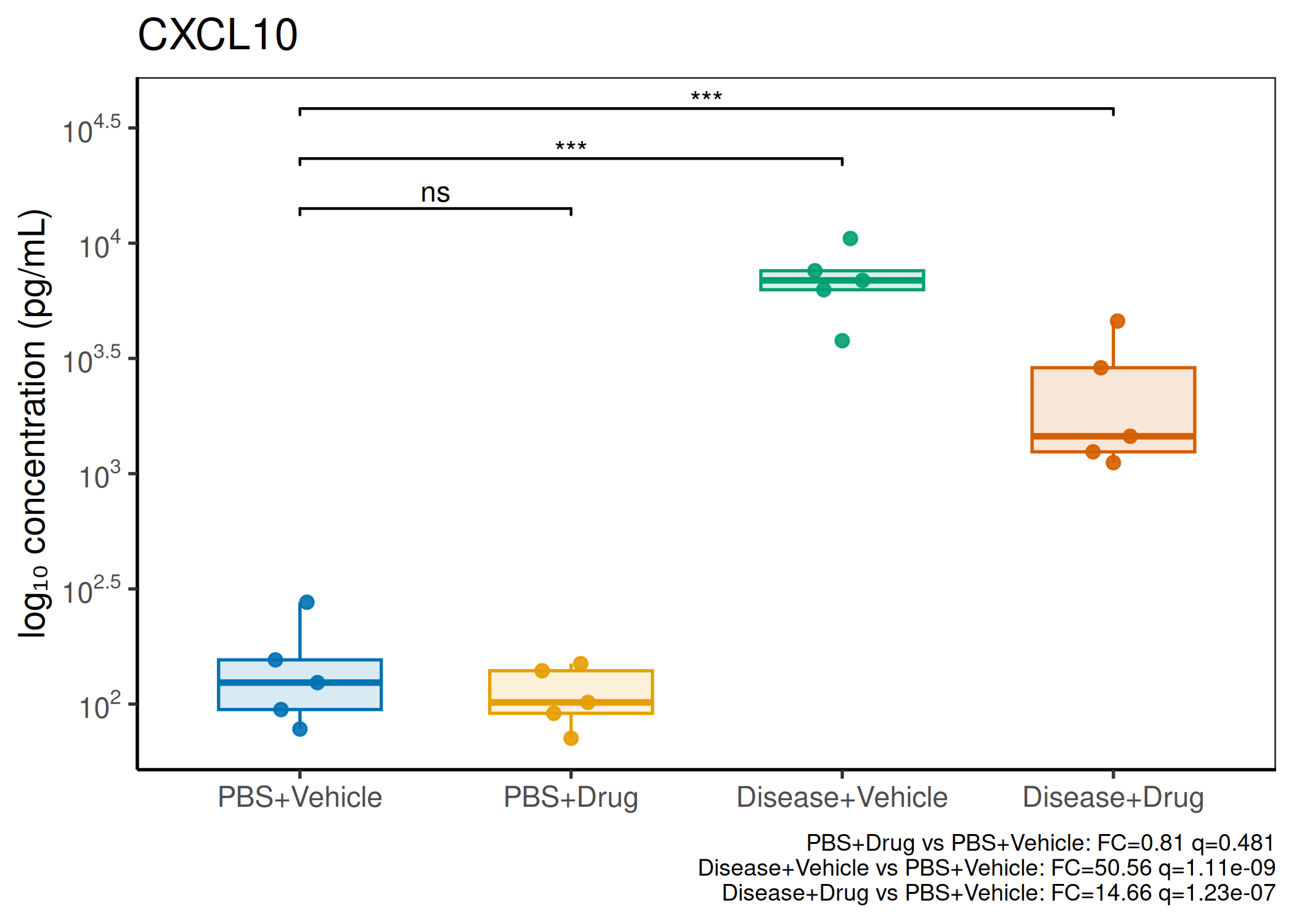
2×2 factorial cytokine analysis with FDR correction and four plot types
immunology
luminex
ANOVA
multiplex
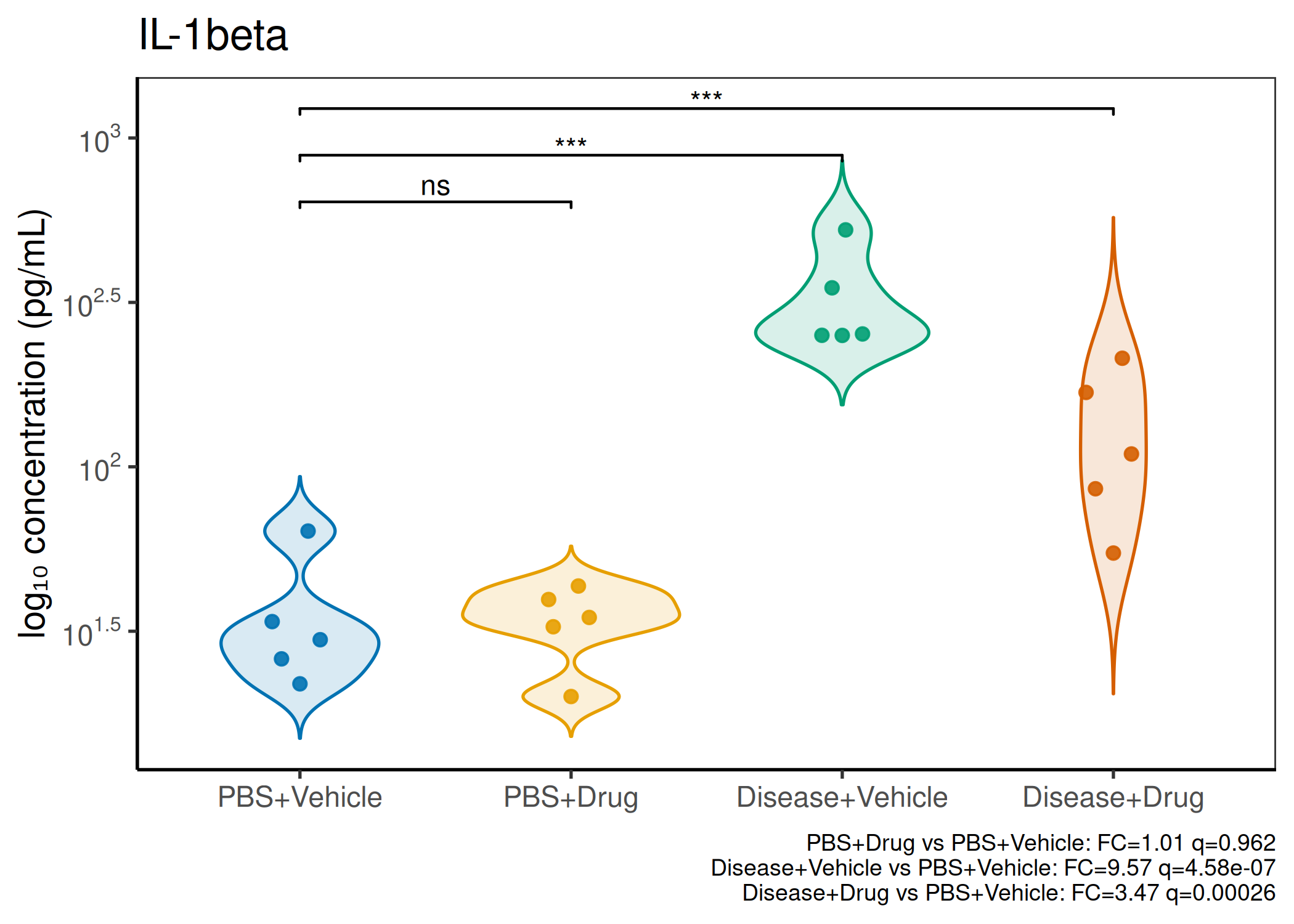
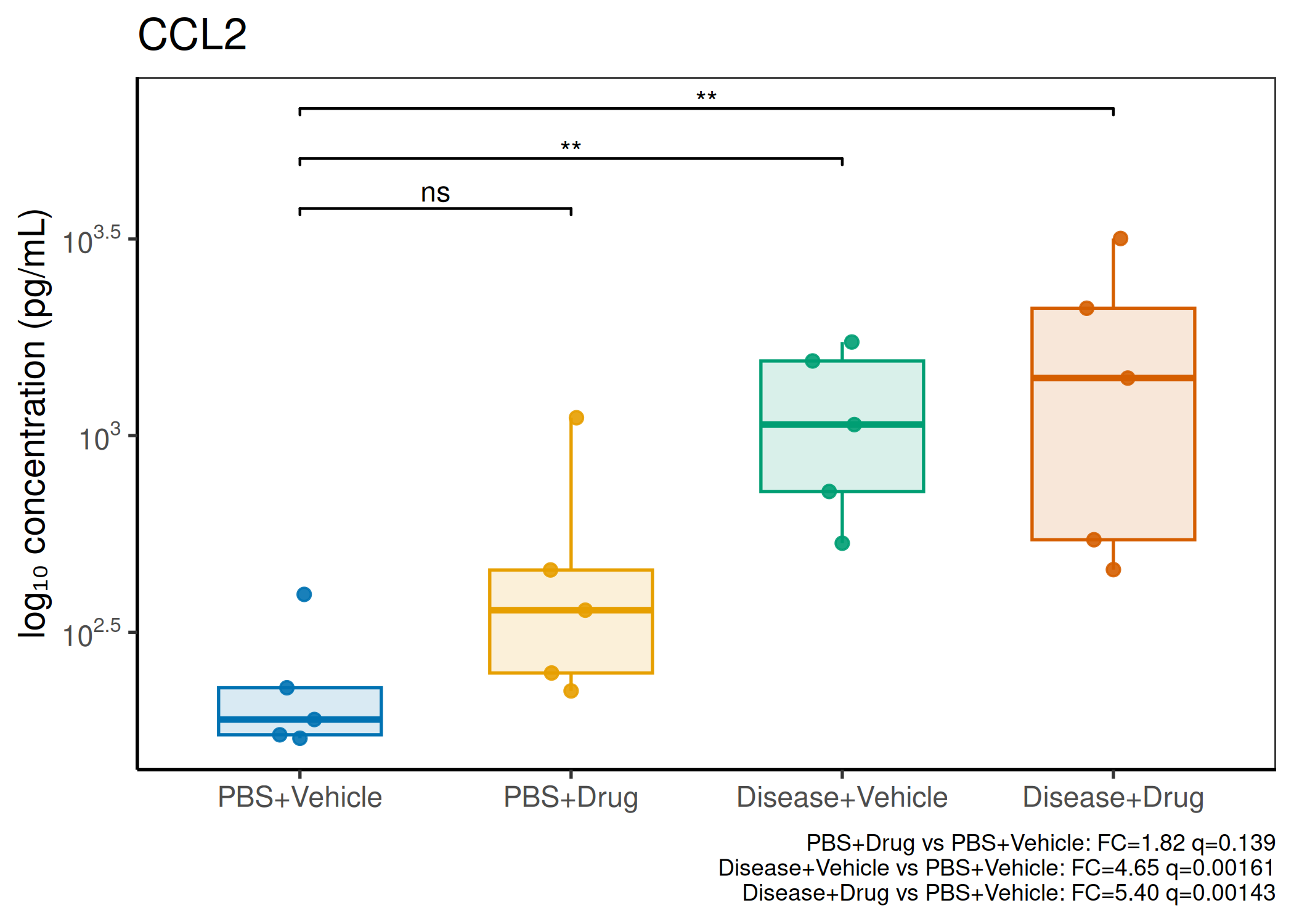
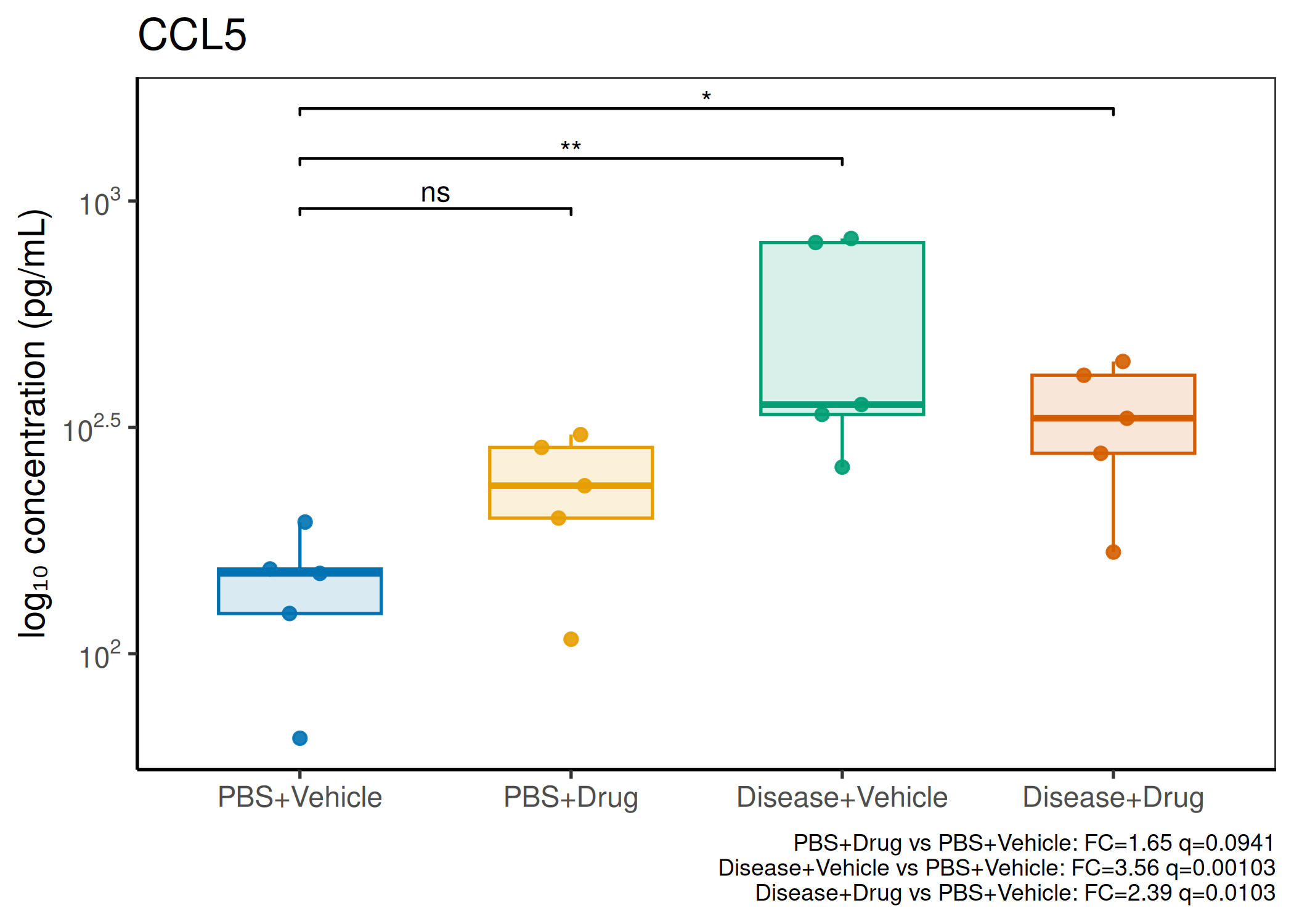
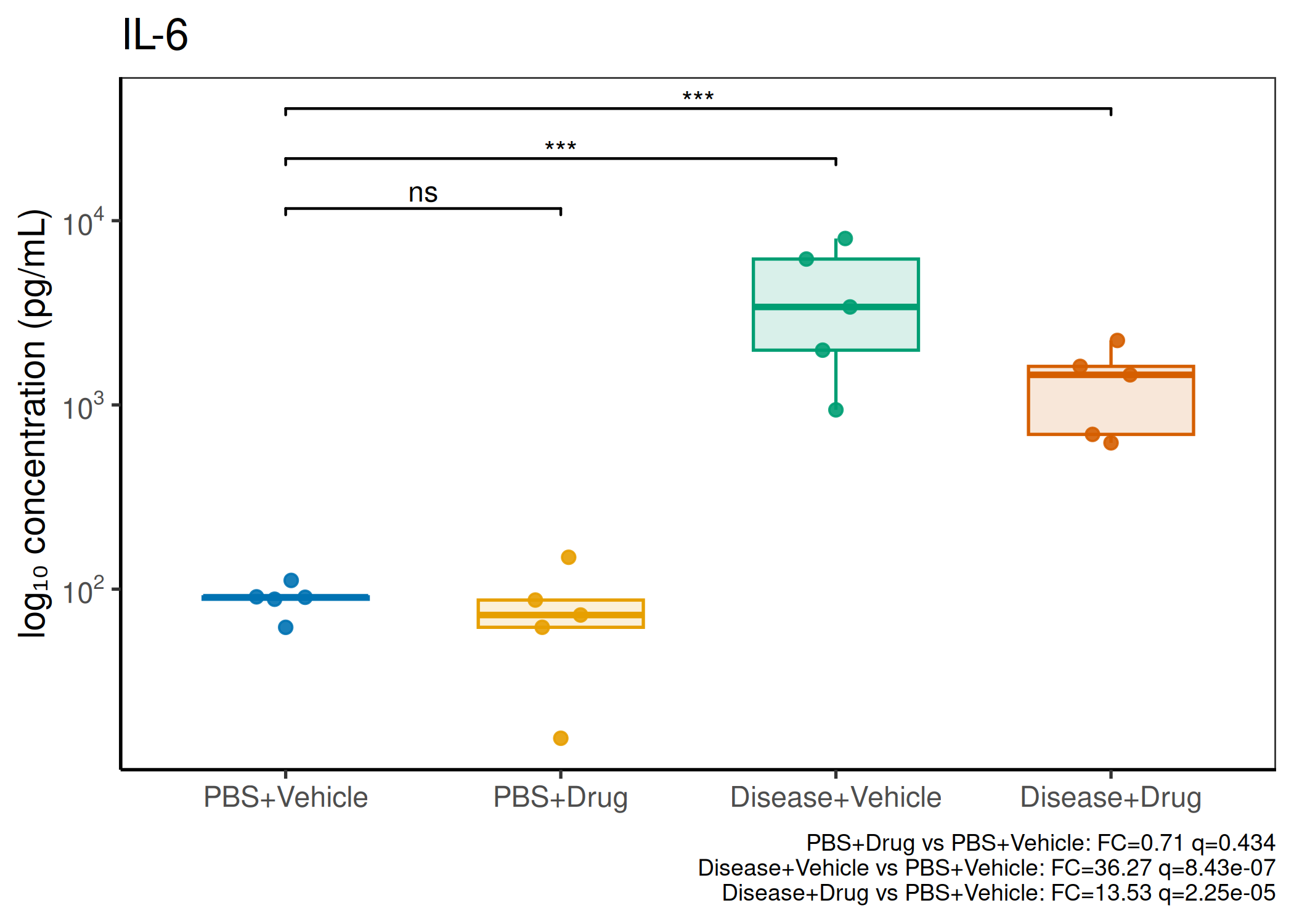
Analyzes multiplex immunoassay (MagPix/Luminex) data from a 2×2 factorial design (e.g., disease × treatment). Handles censored values (< LoD) via LoD/√2 substitution, flags outliers (3×IQR), fits per-analyte two-way ANOVA, computes emmeans contrasts with BH-FDR, and produces a heatmap plus four per-analyte plot types (dot, bar, violin, box).
When to use this template: You have a MagPix/Luminex panel run on a 2×2 factorial experiment (e.g., disease status × treatment). Adapt FACTOR1_LEVELS and FACTOR2_LEVELS to your own factor names.
Note
Okabe-Ito palette is used throughout — colorblind-safe for all four groups.
## ── USER CONFIGURATION ──────────────────────────────────────────────────────## DATA_FILE: Path to your MagPix/Luminex export CSV.# Required columns (in addition to analyte columns):# - "Location" : well identifier (e.g., "A1")# - "Sample" : sample name — used to derive Factor1 and Factor2# - "Original_ID" : original sample ID (can be same as Sample)# - "Total Events" : bead event count (for QC)#DATA_FILE <-"data/luminex_data.csv"# NON_ANALYTE_COLS: Columns that are NOT cytokine measurements.# All other columns will be treated as analytes.NON_ANALYTE_COLS <-c("Location", "Sample", "Original_ID", "Total Events")# FACTOR1_PATTERN / FACTOR2_PATTERN: Regex patterns matched against the# Sample column to assign Factor1 and Factor2.# The default detects "<name>" at start-of-string for Factor1,# and "<name>" anywhere for Factor2.## Example: Sample = "Disease_Drug_1"# FACTOR1_LEVELS[2] = "Disease" → if str_detect(Sample, "Disease") → "Disease"# FACTOR2_LEVELS[2] = "Drug" → if str_detect(Sample, "Drug") → "Drug"#FACTOR1_LEVELS <-c("PBS", "Disease") # [1] = reference / control levelFACTOR2_LEVELS <-c("Vehicle", "Drug") # [1] = reference / control level# BASELINE_GROUP: The reference group label for emmeans contrasts.# Format: "<FACTOR1_ref>+<FACTOR2_ref>"BASELINE_GROUP <-paste0(FACTOR1_LEVELS[1], "+", FACTOR2_LEVELS[1])# OKABE_ITO: Colorblind-safe 4-color palette for the four 2×2 groups.# Names follow the "<Factor1>+<Factor2>" convention.OKABE_ITO <-c("PBS+Vehicle"="#0072B2", # Blue"PBS+Drug"="#E69F00", # Orange"Disease+Vehicle"="#009E73", # Bluish green"Disease+Drug"="#D55E00"# Vermilion)# CONTRAST_ORDER / CONTRAST_LABELS: Define which emmeans contrasts appear in# the heatmap (order matters for the y-axis) and their display labels.# Adjust if your Factor levels produce different contrast names.CONTRAST_ORDER <-c(paste0(FACTOR2_LEVELS[2], " - ", FACTOR2_LEVELS[1], ", ", FACTOR1_LEVELS[1]),paste0(FACTOR2_LEVELS[2], " - ", FACTOR2_LEVELS[1], ", ", FACTOR1_LEVELS[2]),paste0(FACTOR1_LEVELS[2], " - ", FACTOR1_LEVELS[1], ", ", FACTOR2_LEVELS[1]),paste0(FACTOR1_LEVELS[2], " - ", FACTOR1_LEVELS[1], ", ", FACTOR2_LEVELS[2]))CONTRAST_LABELS <-c(paste0("Drug (", FACTOR1_LEVELS[1], ")"),paste0("Drug (", FACTOR1_LEVELS[2], ")"),paste0(FACTOR1_LEVELS[2], " (", FACTOR2_LEVELS[1], ")"),paste0(FACTOR1_LEVELS[2], " (", FACTOR2_LEVELS[2], ")"))# OUTPUT_DIR: Folder where per-analyte plots are saved.OUTPUT_DIR <-"Plots"## ────────────────────────────────────────────────────────────────────────────
MagPix / Luminex Best Practices
Bead counts: ≥35 per analyte (≥50 ideal). Re-export with bead counts if missing.
Censoring (“< LoD”): Do not treat as zero. Replace with LoD/√2 for substitution.
QC/outliers: Flag wells using Tukey 3×IQR on the log10 scale.
Design: 2×2 factorial — model on log10(concentration), test main effects and interaction.
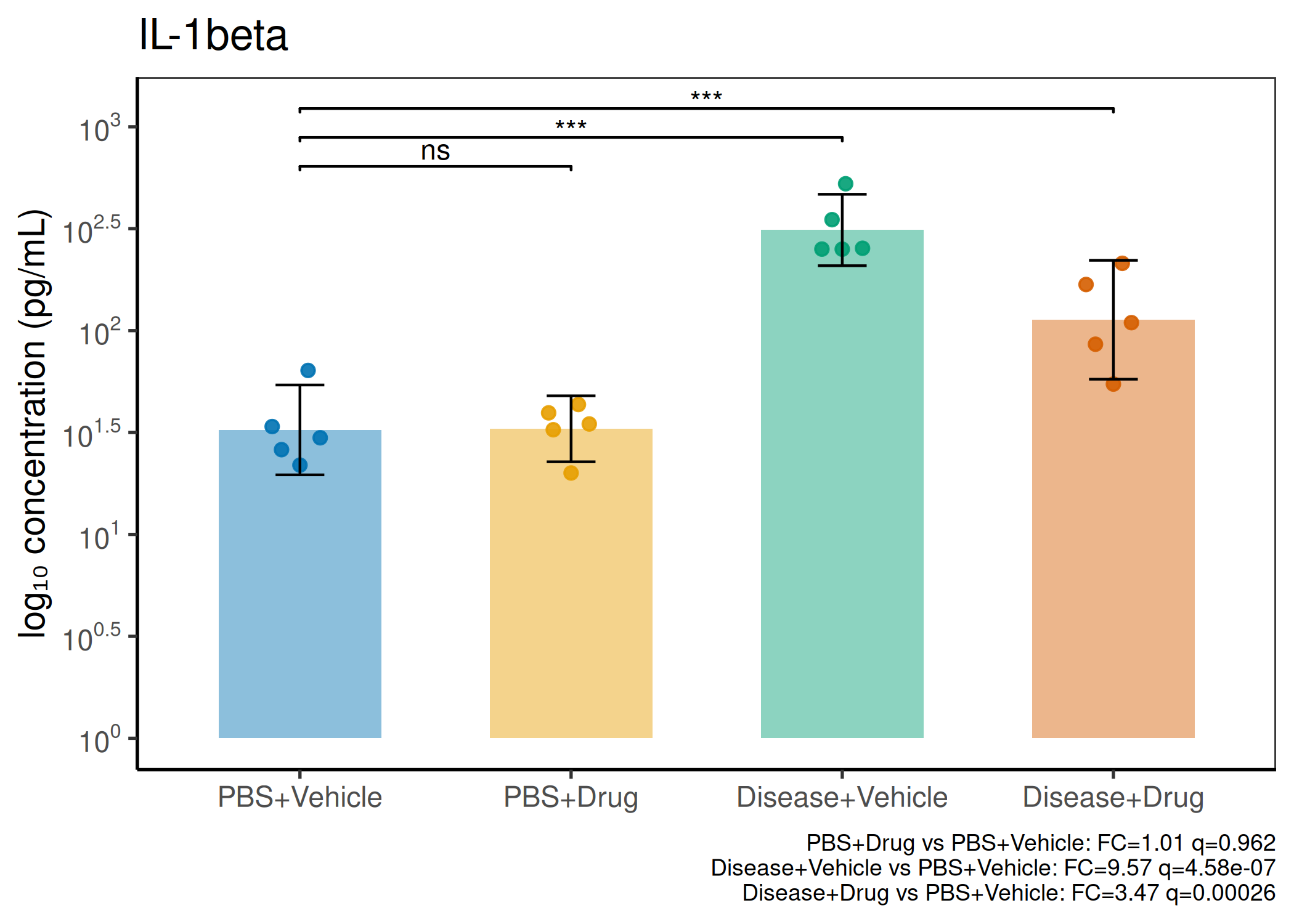
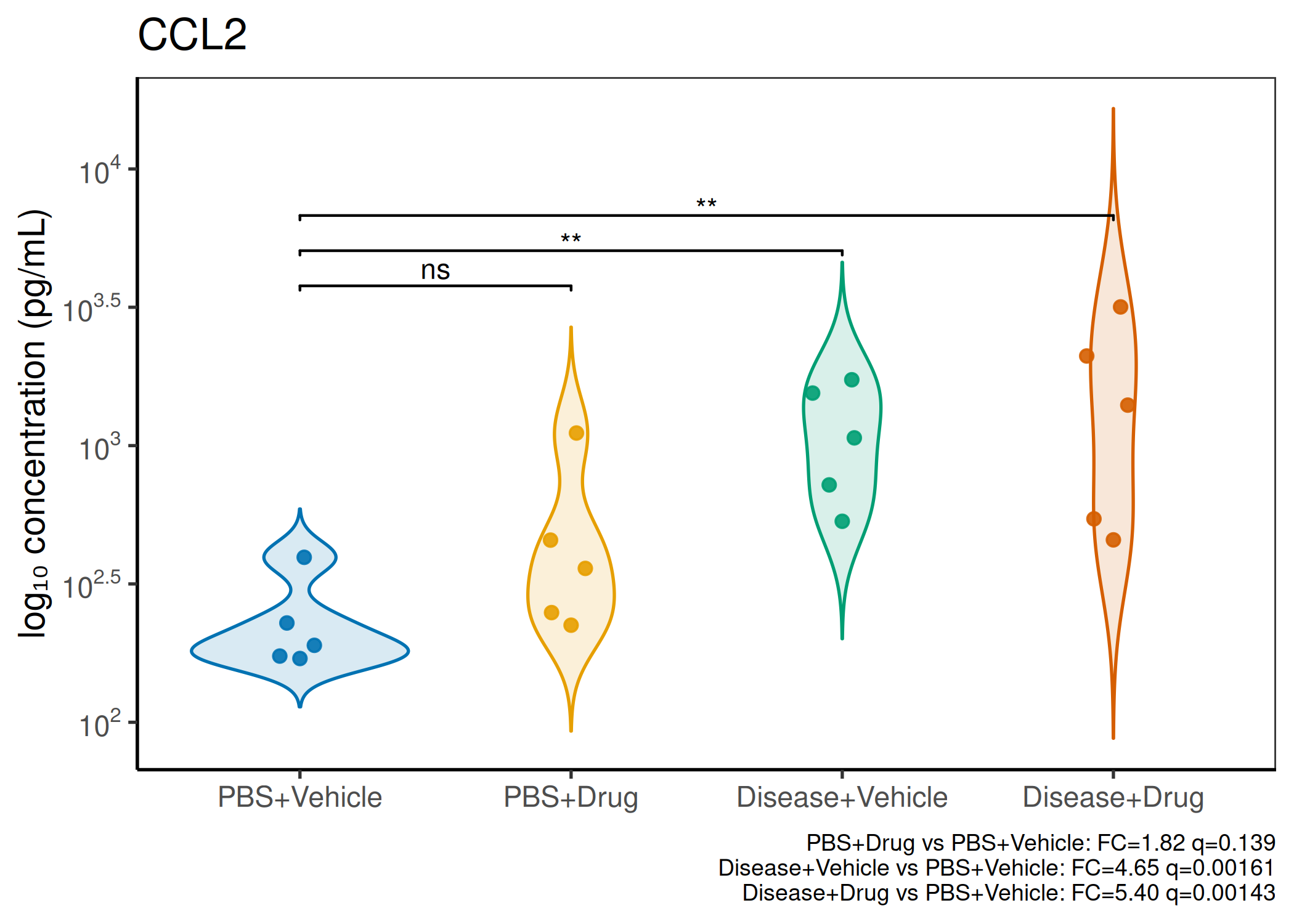
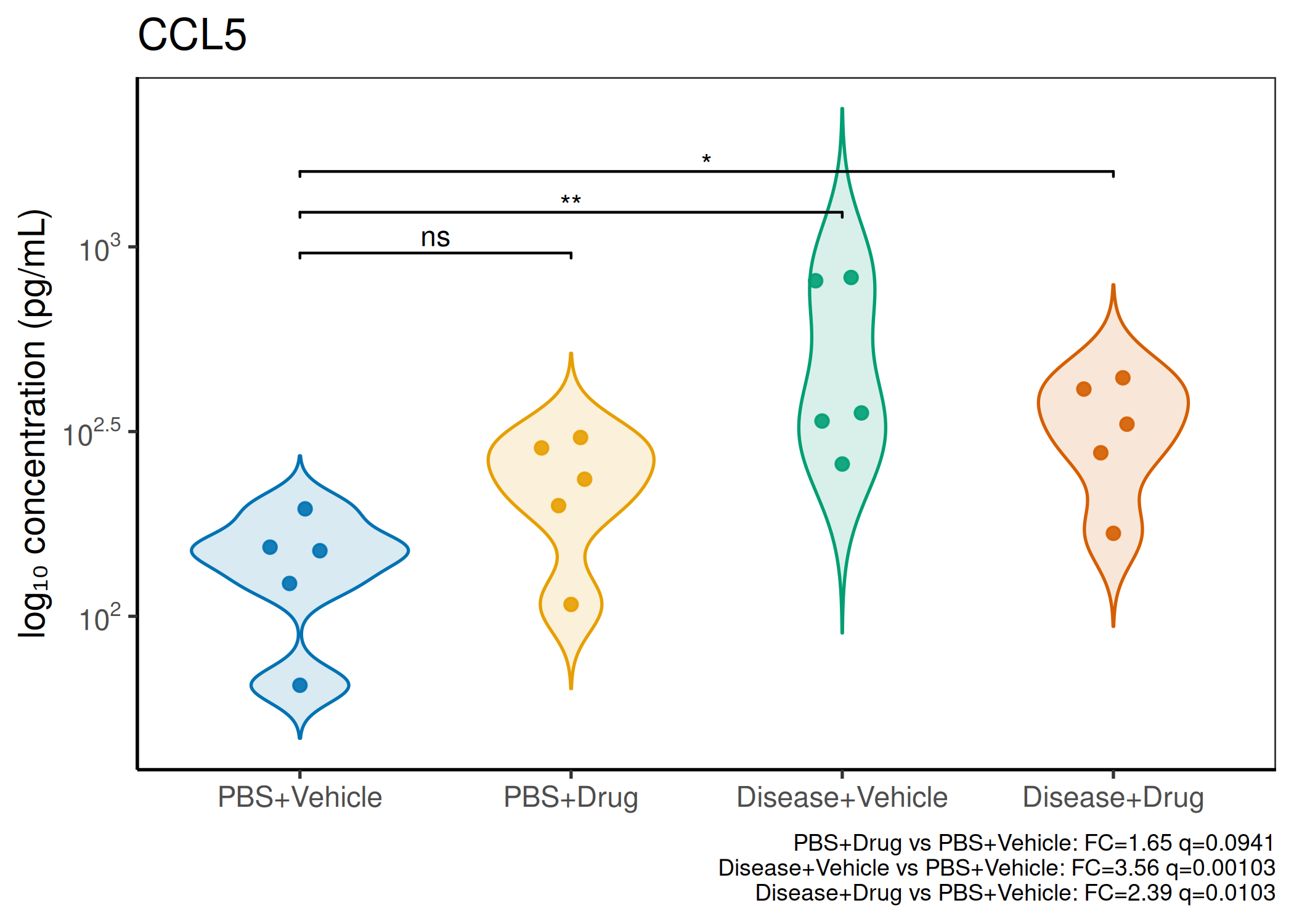
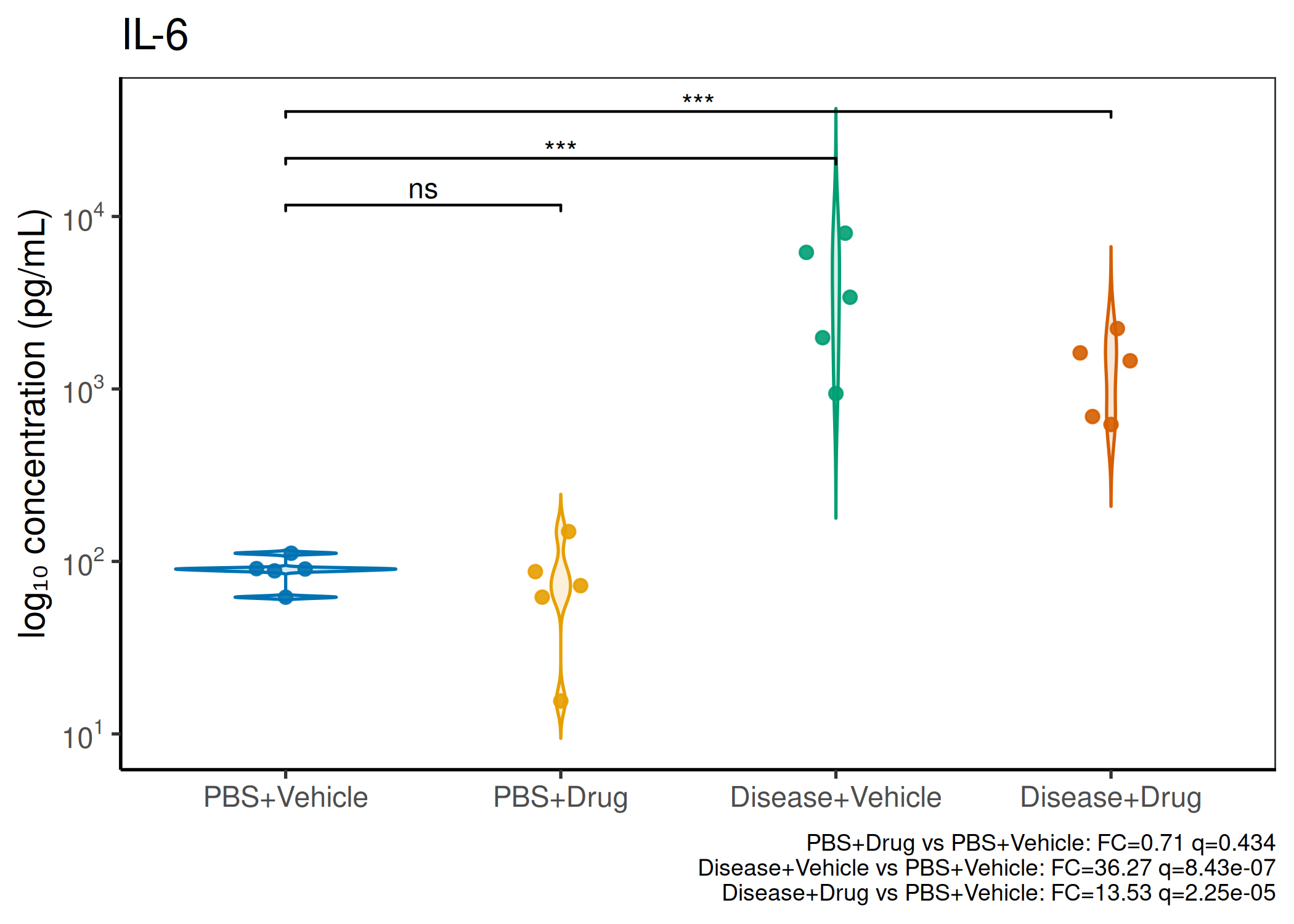
Multiple analytes: Correct FDR across analytes (BH). Report fold-changes as 10^β.
Guidance: Analytes with >50% censoring warrant Tobit regression as a sensitivity check. The standard substitution (LoD/√2) used here is appropriate for <30% censoring.
---title: "04 · MagPix / Luminex Multiplex"subtitle: "2×2 factorial cytokine analysis with FDR correction and four plot types"description: | Analyzes multiplex immunoassay (MagPix/Luminex) data from a 2×2 factorial design (e.g., disease × treatment). Handles censored values (< LoD) via LoD/√2 substitution, flags outliers (3×IQR), fits per-analyte two-way ANOVA, computes emmeans contrasts with BH-FDR, and produces a heatmap plus four per-analyte plot types (dot, bar, violin, box).categories: [immunology, luminex, ANOVA, multiplex]---## Overview| Item | Details ||------|---------|| **Input** | Single wide-format CSV (rows = samples, columns = analytes + metadata) || **Key packages** | `tidyverse`, `emmeans`, `ggbeeswarm`, `ggpubr`, `scales` || **Statistics** | Two-way ANOVA per analyte · emmeans contrasts · BH/FDR || **Output** | QC tables · ANOVA results CSV · Contrasts CSV · Heatmap · 4 plot types per analyte || **Download** | [template.Rmd](template.Rmd) |::: {.callout-tip}**When to use this template:** You have a MagPix/Luminex panel run on a 2×2 factorialexperiment (e.g., disease status × treatment). Adapt `FACTOR1_LEVELS` and`FACTOR2_LEVELS` to your own factor names.:::::: {.callout-note}**Okabe-Ito palette** is used throughout — colorblind-safe for all four groups.:::[Back to Gallery](../../index.html){.btn .btn-outline-secondary}[Open Template File](template.Rmd){.btn .btn-primary}```{r child="template.Rmd"}```