Isolation by Distance (Mantel Test), Scatter Pie Map, and Mantel Correlogram
phylogenetics
geographic
mantel
virology
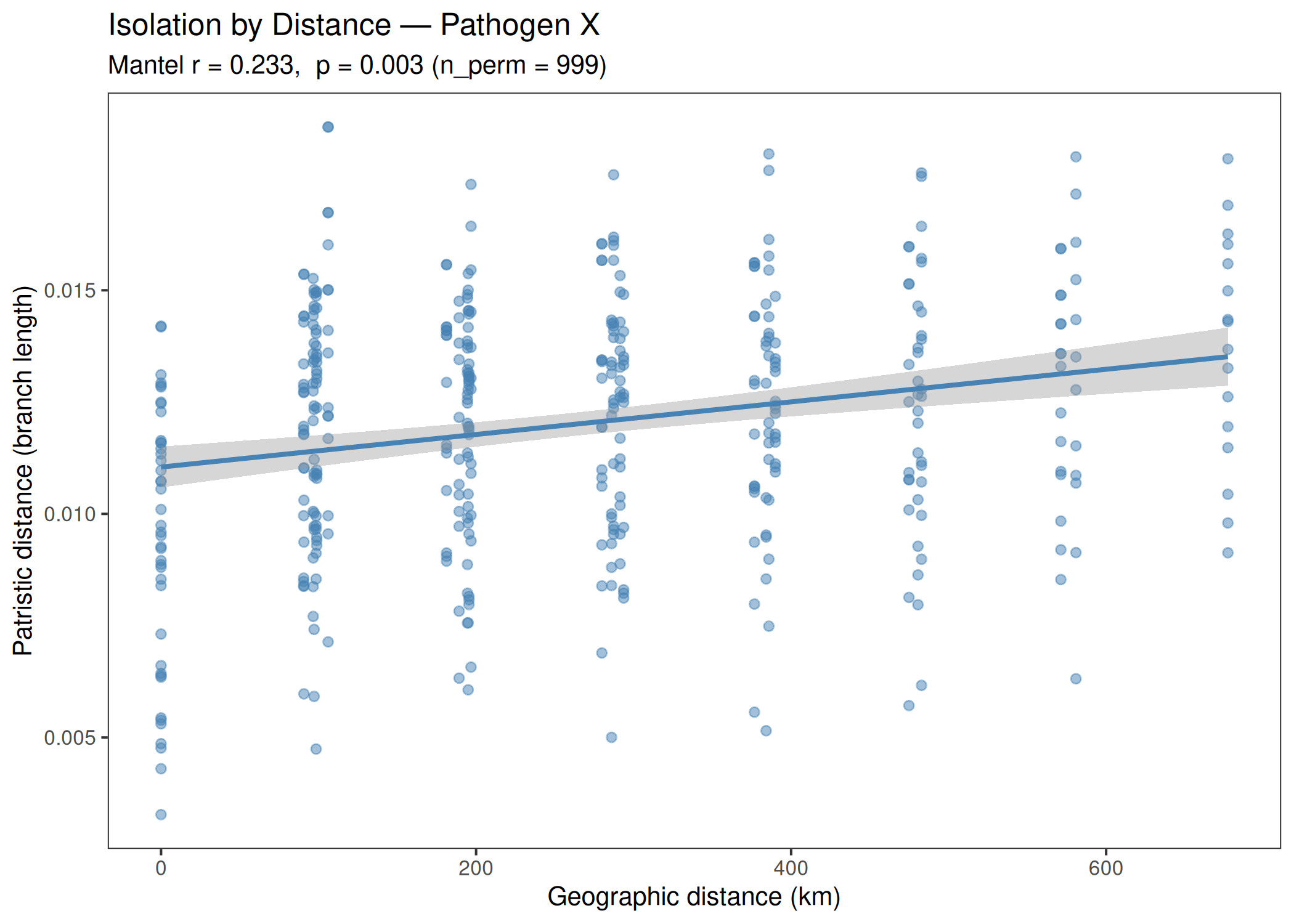
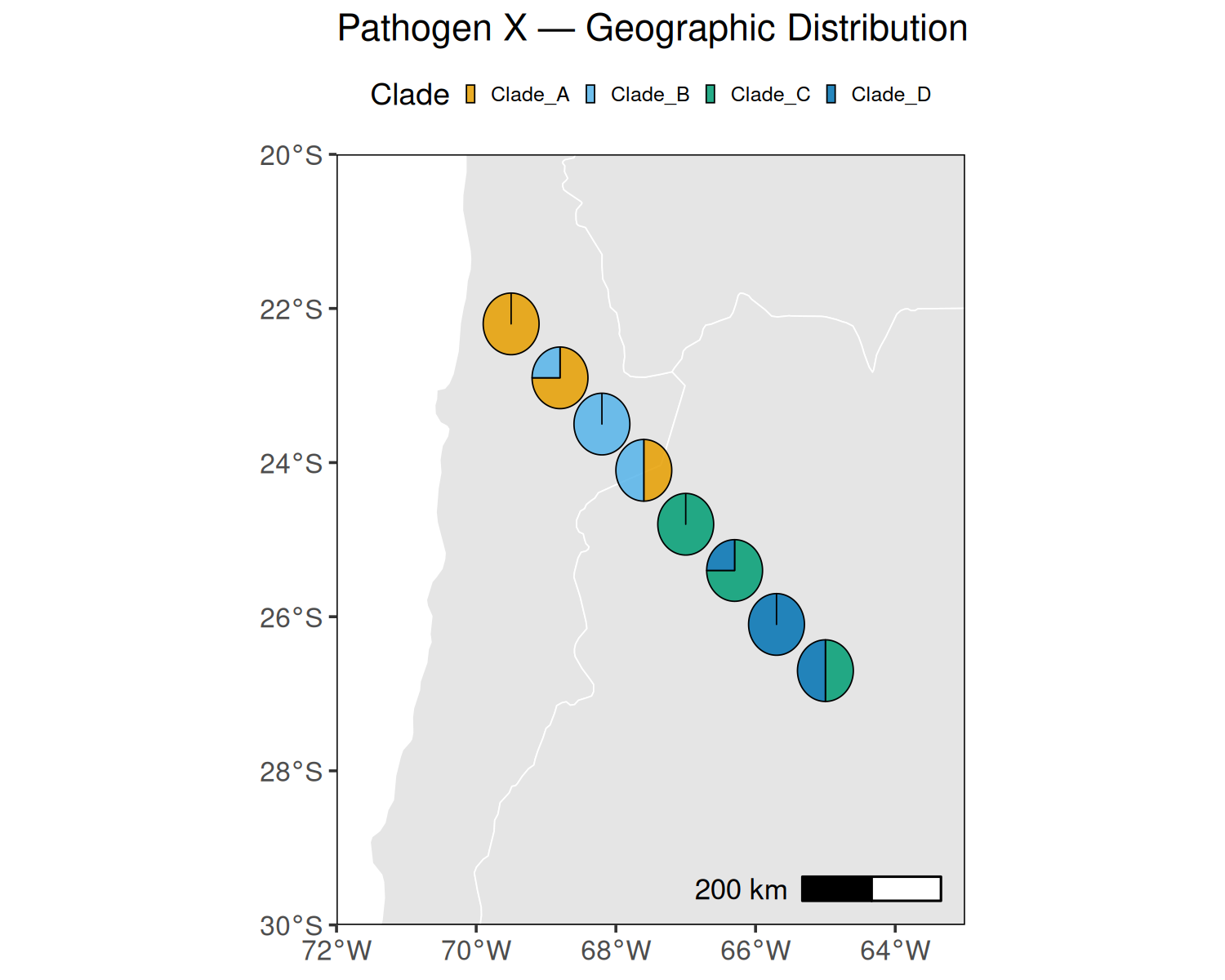
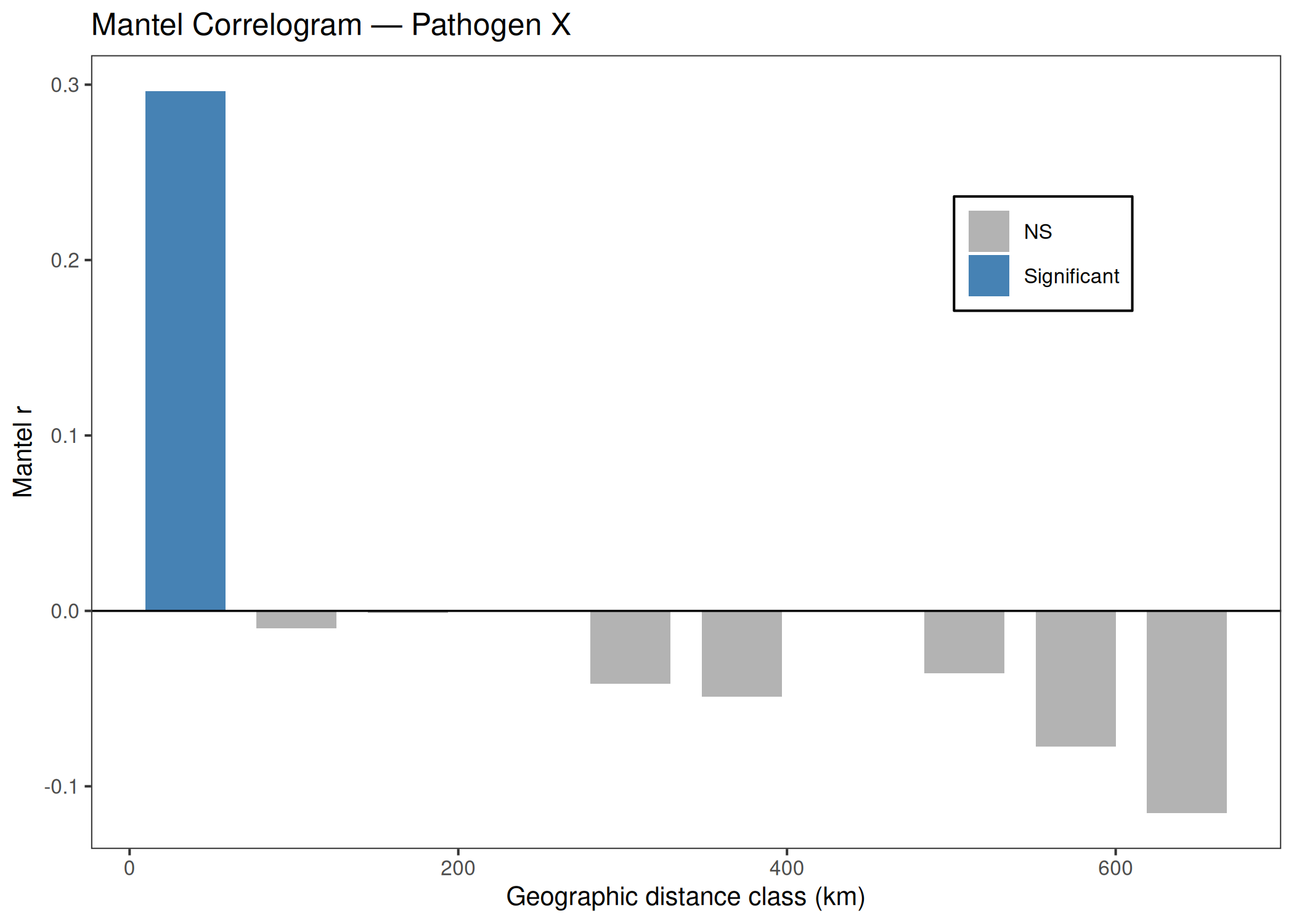
Performs isolation-by-distance analysis using Mantel tests on patristic distances from a phylogenetic tree and geographic (Haversine) distances. Produces a scatter pie map showing clade composition per sampling location and a Mantel correlogram.
Overview
Item
Details
Input
Aligned FASTA (S segment required, M optional) · Newick tree · coordinates CSV · metadata CSV
When to use this template: You have a phylogenetic tree and sampling coordinates for a pathogen, and want to test whether genetic distance correlates with geographic distance (isolation by distance) and visualize the geographic distribution of clades.
Note
Clade colors: Default palette uses Okabe-Ito colorblind-safe colors. Update CLADE_COLORS in the USER CONFIGURATION block to match your clade assignments. The scatter pie radius (radius column in pie_data) is in degrees — adjust mutate(radius = 0.4) to scale pies appropriately for your map extent.
## ── USER CONFIGURATION ──────────────────────────────────────────────────────## Input files:S_FASTA <-"data/S_aligned.fasta"# Aligned S-segment FASTAM_FASTA <-"data/M_aligned.fasta"# Aligned M-segment FASTA (optional)TREEFILE <-"data/S_tree.nwk"# Newick tree from IQ-TREE or similarCOORDS_FILE<-"data/coordinates.csv"# Columns: SampleID, Longitude, LatitudeMETADATA_FILE <-"data/metadata.csv"# Columns: SampleID, Clade (+ optional extras)# Column names in COORDS_FILE and METADATA_FILE that link to FASTA tip labels.SAMPLE_COL <-"SampleID"# Columns in METADATA_FILE used for clade coloring.CLADE_COL <-"Clade"# CLADE_COLORS: Named color vector (Okabe-Ito palette).# Names must match the unique values in METADATA_FILE[[CLADE_COL]].CLADE_COLORS <-c("Clade_A"="#E69F00", # Orange"Clade_B"="#56B4E9", # Sky blue"Clade_C"="#009E73", # Bluish green"Clade_D"="#0072B2"# Blue)# PATHOGEN_NAME: Used in plot titles and axis labels.PATHOGEN_NAME <-"Pathogen X"# Mantel test: number of permutations (increase to 99999 for publication).MANTEL_NPERM <-999# Map extent (degrees): lon/lat range around your sampling area.MAP_LON <-c(-72, -63)MAP_LAT <-c(-30, -20)# OUTPUT_DIR: Where to save plots.OUTPUT_DIR <-"Plots"## ────────────────────────────────────────────────────────────────────────────
# ── Summarize clade composition per location ───────────────────────────────────# (when multiple samples share a location, show their clade proportions as a pie)pie_data <- sample_data %>%group_by(Longitude, Latitude, !!sym(CLADE_COL)) %>%summarise(n =n(), .groups ="drop") %>% tidyr::pivot_wider(names_from =all_of(CLADE_COL), values_from = n, values_fill =0) %>%mutate(radius =0.4) # pie radius in degrees; adjust to your map scale# Country polygonsworld <-ne_countries(scale ="medium", returnclass ="sf")p_spie <-ggplot() +geom_sf(data = world, fill ="grey90", color ="white", linewidth =0.2) +geom_scatterpie(data = pie_data,aes(x = Longitude, y = Latitude, r = radius),cols =intersect(names(CLADE_COLORS), names(pie_data)),alpha =0.85, color ="black", linewidth =0.2 ) +scale_fill_manual(values = CLADE_COLORS, name ="Clade") +coord_sf(xlim = MAP_LON, ylim = MAP_LAT, expand =FALSE) +annotation_scale(location ="br", width_hint =0.25) +labs(title =paste(PATHOGEN_NAME, "— Geographic Distribution"),x =NULL, y =NULL) +theme_bw(base_size =9, base_family ="sans") +theme(legend.position ="top",legend.direction ="horizontal",legend.text =element_text(size =6),legend.key.size =unit(3, "pt"),panel.grid.major =element_blank(),panel.grid.minor =element_blank(),panel.border =element_rect(color ="black", linewidth =0.35),axis.text =element_text(size =8),plot.margin =margin(4, 4, 4, 4, "pt") )ggsave(file.path(OUTPUT_DIR, "Maps", "scatter_pie.tiff"),plot = p_spie, width =3, height =2, dpi =600,device ="tiff", compression ="lzw")ggsave(file.path(OUTPUT_DIR, "Maps", "scatter_pie.svg"),plot = p_spie, width =3, height =2, device ="svg")print(p_spie)
Mantel Correlogram
Code
# Distance class boundaries (km) — adjust to your study areadist_breaks <-quantile(as.vector(geo_sub[lower.tri(geo_sub)]),probs =seq(0, 1, by =0.25))# Mantel correlogram (vegan)mcor <-mantel.correlog(D.eco = pat_sub,D.geo = geo_sub,nperm = MANTEL_NPERM,cutoff =FALSE)mcor_df <-as.data.frame(mcor$mantel.res, check.names =FALSE)# vegan reports raw and, when mult != "none", corrected p-values.# Match the package's own plotting behavior by preferring corrected p-values.p_col <-if ("Pr(corrected)"%in%names(mcor_df)) "Pr(corrected)"else"Pr(Mantel)"mcor_df <- mcor_df %>%mutate(pvalue = .data[[p_col]],sig =ifelse(!is.na(pvalue) & pvalue <=0.05, "Significant", "NS") )p_mcor <-ggplot(mcor_df, aes(x =`class.index`, y = Mantel.cor, fill = sig)) +geom_bar(stat ="identity", width =diff(range(mcor_df$`class.index`)) /nrow(mcor_df) *0.8) +geom_hline(yintercept =0, color ="black", linewidth =0.4) +scale_fill_manual(values =c("Significant"="steelblue", "NS"="grey70"),name =NULL) +labs(title =paste("Mantel Correlogram —", PATHOGEN_NAME),x ="Geographic distance class (km)",y ="Mantel r") +theme_bw(base_size =10, base_family ="sans") +theme(legend.position =c(0.8, 0.75),legend.background =element_rect(color ="black"),legend.key =element_rect(fill ="transparent"),panel.grid.major =element_blank(),panel.grid.minor =element_blank() )ggsave(file.path(OUTPUT_DIR, "Mantel", "mantel_correlogram.tiff"),plot = p_mcor, width =3, height =2, dpi =300,device ="tiff", compression ="lzw")print(p_mcor)
Source Code
---title: "05 · Phylo-Geographic Analysis"subtitle: "Isolation by Distance (Mantel Test), Scatter Pie Map, and Mantel Correlogram"description: | Performs isolation-by-distance analysis using Mantel tests on patristic distances from a phylogenetic tree and geographic (Haversine) distances. Produces a scatter pie map showing clade composition per sampling location and a Mantel correlogram.categories: [phylogenetics, geographic, mantel, virology]---## Overview| Item | Details ||------|---------|| **Input** | Aligned FASTA (S segment required, M optional) · Newick tree · coordinates CSV · metadata CSV || **Key packages** | `ape`, `vegan`, `geosphere`, `scatterpie`, `sf`, `rnaturalearth`, `ggspatial` || **Statistics** | Mantel test (Pearson) · patristic distances · Haversine geographic distances || **Output** | Isolation-by-distance plot · scatter pie map · Mantel correlogram · TIFF + SVG || **Download** | [template.Rmd](template.Rmd) |::: {.callout-tip}**When to use this template:** You have a phylogenetic tree and sampling coordinatesfor a pathogen, and want to test whether genetic distance correlates with geographicdistance (isolation by distance) and visualize the geographic distribution of clades.:::::: {.callout-note}**Clade colors:** Default palette uses Okabe-Ito colorblind-safe colors.Update `CLADE_COLORS` in the USER CONFIGURATION block to match your clade assignments.The scatter pie radius (`radius` column in `pie_data`) is in degrees — adjust `mutate(radius = 0.4)`to scale pies appropriately for your map extent.:::[Back to Gallery](../../index.html){.btn .btn-outline-secondary}[Open Template File](template.Rmd){.btn .btn-primary}```{r child="template.Rmd"}```